Unlocking Nature's Medicine Chest: A Cutting-Edge Guide to PKS Gene Splitting for Enhanced Biosynthesis
This comprehensive guide provides researchers and drug development professionals with a strategic framework for leveraging polyketide synthase (PKS) gene splitting to overcome key bottlenecks in natural product biosynthesis.
Unlocking Nature's Medicine Chest: A Cutting-Edge Guide to PKS Gene Splitting for Enhanced Biosynthesis
Abstract
This comprehensive guide provides researchers and drug development professionals with a strategic framework for leveraging polyketide synthase (PKS) gene splitting to overcome key bottlenecks in natural product biosynthesis. We cover the foundational principles of PKS megaenzyme architecture and the rationale for splitting, delve into practical methodologies for split-site selection and heterologous expression, address common troubleshooting and optimization challenges, and validate the approach through comparative analysis with traditional methods. This article synthesizes the latest advancements to empower the efficient microbial production of high-value pharmaceuticals and biomolecules.
Deconstructing the Megaenzyme: Foundational Principles of PKS Architecture and Split-Site Rationale
Application Notes: PKSs in Drug Discovery and Engineering
Polyketide synthases (PKSs) are multi-domain megasynthetases that assemble complex natural products with diverse bioactivities, including antibiotics (erythromycin), antifungals (amphotericin), and anticancer agents (epothilone). Their modular, assembly-line logic makes them prime targets for bioengineering to produce novel therapeutics. Recent advancements, particularly in PKS gene splitting strategies, are overcoming historical challenges in manipulating these large, complex systems for improved biosynthesis.
Table 1: Representative Polyketide Drugs and Their PKS Types
| Drug | Therapeutic Class | PKS Type (I, II, III) | Number of Modules* | Key Producing Organism |
|---|---|---|---|---|
| Erythromycin A | Macrolide antibiotic | Type I (Modular) | 6 | Saccharopolyspora erythraea |
| Doxorubicin | Anthracycline anticancer | Type II (Iterative) | 1 (Iterative) | Streptomyces peucetius |
| Tetracycline | Broad-spectrum antibiotic | Type II (Iterative) | 1 (Iterative) | Streptomyces aureofaciens |
| Epothilone B | Microtubule stabilizer | Type I (Modular) | 9 | Sorangium cellulosum |
| Lovastatin | Cholesterol-lowering | Type I (Iterative) | 2 (Iterative) | Aspergillus terreus |
*For Type I modular systems.
Core Thesis Context: The Gene Splitting Strategy A central thesis in modern PKS engineering posits that splitting large, contiguous PKS genes into discrete, manageable expression cassettes (a "split-PKS" approach) significantly improves biosynthetic titers and enables precise module swapping. This strategy addresses issues of genetic instability, poor heterologous expression, and inefficient protein folding associated with mega-gene clusters. It facilitates the construction of optimized chimeric PKSs for combinatorial biosynthesis.
Experimental Protocols
Protocol 2.1: Heterologous Expression of a Split Type I PKS Module inStreptomyces coelicolor
Objective: To express and assess the activity of a single PKS module (e.g., Module 3 of the 6-deoxyerythronolide B synthase, DEBS) from Saccharopolyspora erythraea after splitting it from the native polycistron.
Materials (Research Reagent Solutions):
- pSET152-derived Expression Vector: Streptomyces-E. coli shuttle vector with constitutive ermEp promoter and apramycin resistance (aac(3)IV).
- Gateway BP/LR Clonase Enzyme Mix: For efficient, site-specific recombination of the split PKS gene fragment into the destination vector.
- S. coelicolor M1154: Engineered heterologous host with deleted endogenous PKS clusters and enhanced precursor supply.
- Tris-HCl Buffered Saline (TBS, 50 mM, pH 7.5): For protein extraction and washing.
- Ni-NTA Agarose Resin: For affinity purification of His-tagged PKS proteins.
- S-Adenosyl Methionine (SAM, 1 mM): Methyl donor for methyltransferase domain assays.
- [1-¹⁴C] Methylmalonyl-CoA: Radiolabeled extender unit for in vitro activity assays.
- Native PAGE Gel (3-8%): For analyzing the assembly and size of intact PKS multienzyme complexes.
Methodology:
- Gene Design & Synthesis: Design a DNA fragment encoding DEBS Module 3 (KS-AT-DH-KR-ACP). Flank it with attB sites for Gateway cloning. Codon-optimize for S. coelicolor and synthesize.
- Gateway Cloning: Perform a BP recombination reaction between the attB-flanked gene and a pDONR221 vector to create an Entry Clone. Sequence-verify. Perform an LR recombination with the destination expression vector pXH7 (derived from pSET152, containing an N-terminal His₆-tag and ermEp).
- Conjugal Transfer to Streptomyces: a. Transform the expression construct into E. coli ET12567/pUZ8002. b. Co-cultivate with S. coelicolor M1154 spores on SFM agar plates at 30°C for 16h. c. Overlay with apramycin (50 µg/mL) and nalidixic acid (25 µg/mL). Incubate until exconjugants appear (5-7 days).
- Fermentation & Protein Extraction: a. Inoculate exconjugants into TSB + apramycin medium. Grow at 30°C, 250 rpm for 48h. b. Harvest cells by centrifugation (4,000 x g, 15 min). Resuspend pellet in 5 mL/g lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 10% glycerol, 1 mM PMSF). c. Lyse cells by sonication (10 cycles of 30s on/30s off, 50% amplitude). Clarify by centrifugation (15,000 x g, 45 min, 4°C).
- Affinity Purification: Incubate supernatant with 1 mL Ni-NTA resin for 1h at 4°C. Wash with 20 column volumes (CV) of wash buffer (lysis buffer + 20 mM imidazole). Elute with elution buffer (lysis buffer + 250 mM imidazole). Analyze purity by SDS-PAGE.
- In Vitro Activity Assay: a. Assemble a 100 µL reaction: 50 mM Tris-HCl (pH 7.5), 2 mM DTT, 5 µM purified PKS module, 100 µM N-acetylcysteamine (SNAC) thioester of the diketide substrate [(2S,3R)-2-methyl-3-hydroxypentanoyl-SNAC], 100 µM [1-¹⁴C]methylmalonyl-CoA, 5 mM MgCl₂. b. Incubate at 30°C for 1h. Quench with 20 µL glacial acetic acid. c. Extract with ethyl acetate (2 x 200 µL). Analyze the organic phase by radio-TLC (Silica Gel 60 F₂₅₄ plate; mobile phase: 19:1 CH₂Cl₂:CH₃OH).
Protocol 2.2: In Vivo Analysis of a Split PKS Pathway via LC-MS Metabolite Profiling
Objective: To detect and quantify novel polyketide intermediates/products from a engineered split-PKS strain.
Methodology:
- Culture Extraction: Grow engineered S. coelicolor strain in 50 mL production medium (e.g., R5 or YEME) for 5-7 days. Centrifuge culture. Separate supernatant and cell pellet.
- Metabolite Extraction: a. Supernatant: Adjust pH to ~3 with 1M HCl. Extract twice with equal volume of ethyl acetate. Dry organic layers in vacuo. b. Pellet: Resuspend in 10 mL methanol, vortex for 30 min, sonicate for 15 min. Centrifuge. Dry supernatant in vacuo.
- LC-MS Analysis: a. Reconstitute dried extracts in 200 µL methanol. b. HPLC Conditions: Column: C18 (2.1 x 100 mm, 1.7 µm). Gradient: 5-95% acetonitrile in water (+0.1% formic acid) over 20 min. Flow: 0.3 mL/min. c. MS Conditions: ESI source in positive/negative mode. Full scan m/z 100-1500. Data-dependent MS/MS on top 5 ions.
Visualizations
Title: Gene Splitting Strategy Workflow
Title: Split-PKS Experimental Protocol Flow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for PKS Gene Splitting and Analysis
| Item | Function in Research | Example/Notes |
|---|---|---|
| Gateway Cloning System | Enables rapid, recombinational cloning of large, split PKS gene fragments into multiple expression hosts. | Thermo Fisher Scientific; Uses attB/attP site-specific recombination. |
| S. coelicolor M1154 Host | An engineered Streptomyces host with deleted endogenous PKS genes, optimized for heterologous expression of actinomycete-derived PKS clusters. | Genetically minimal host reduces background metabolites. |
| pSET152 / pIJ10257 Vectors | Streptomyces-E. coli shuttle vectors with integrating elements (attP-int φC31) for stable chromosomal insertion of PKS genes. | Contain strong, constitutive promoters (ermEp). |
| Ni-NTA Affinity Resin | Purifies His-tagged PKS proteins for in vitro biochemical characterization and structural studies. | Compatible with denaturing or native conditions. |
| SNAC (N-Acetylcysteamine) Thioesters | Soluble, simplified substrate analogs for in vitro PKS activity assays, mimicking the native acyl carrier protein (ACP)-bound state. | Allows measurement of individual module kinetics. |
| Radiolabeled Extender Units (e.g., [¹⁴C]-Malonyl-CoA) | Tracer for highly sensitive detection of polyketide chain extension activity in crude lysates or purified enzyme assays. | Detected via radio-TLC or scintillation counting. |
| Native PAGE Gels (3-8%) | Analyzes the intact quaternary structure and assembly of multimodular PKS proteins without denaturation. | Critical for confirming proper complex formation post-splitting. |
| LC-HRMS System | Provides high-resolution mass detection for identifying novel polyketide structures from engineered split-PKS strains. | Q-TOF or Orbitrap platforms enable precise mass determination. |
Polyketide synthases (PKSs) are colossal multi-enzymatic assembly lines responsible for producing diverse bioactive molecules. Their heterologous expression in tractable hosts like E. coli or S. cerevisiae is critical for pathway engineering and drug production. However, the massive size and complexity of PKS genes present formidable bottlenecks. The quantitative data below summarizes the core challenges.
Table 1: Quantitative Challenges in Heterologous PKS Expression
| Challenge Category | Representative Data/Scale | Consequence for Heterologous Expression |
|---|---|---|
| Gene Size | Type I PKS genes: 10 - 150+ kb. Single module: 3-6 kb. | Exceeds capacity of standard cloning vectors (e.g., plasmids typically <15 kb). |
| GC Content | Often >70% (e.g., from Actinobacteria). | Causes ribosomal stalling, codon bias, mRNA secondary structure, and truncated proteins in hosts like E. coli. |
| Repetitive Sequences | High sequence identity between ketosynthase (KS) and acyltransferase (AT) domains across modules. | Promotes homologous recombination in vivo, leading to gene deletion and rearrangement. |
| Protein Size | Multi-domain polypeptides: 100 - 10,000+ kDa. | Challenges cellular folding machinery, leads to aggregation, inclusion bodies, and low soluble yield. |
| Codon Bias | Rare codon frequency >30% in high-GC genes for E. coli. | Depletes charged tRNA pools, drastically reduces translation efficiency and protein fidelity. |
| Host Toxicity | Production of reactive intermediates or membrane disruption. | Host cell growth inhibition, low biomass, and failure to sustain pathway expression. |
Detailed Experimental Protocols
Protocol 2.1: Assessing PKS Gene Expression and Solubility inE. coli
Objective: To evaluate the initial expression potential and solubility of a large PKS gene segment in a heterologous host.
Materials:
- Expression Vector: pET-28a(+) (or similar with T7/lac promoter, His-tag).
- Host Strain: E. coli BL21(DE3) pLysS (for tight control) and E. coli Origami 2(DE3) (enhanced disulfide bond formation).
- PKS Gene Fragment: Codon-optimized synthetic fragment (5-8 kb) cloned into vector.
- Media: LB broth with appropriate antibiotics (Kanamycin, Chloramphenicol).
- Inducer: Isopropyl β-D-1-thiogalactopyranoside (IPTG).
Procedure:
- Transformation: Transform the PKS construct into both E. coli expression strains via heat shock or electroporation. Plate on selective LB agar.
- Small-scale Induction: Inoculate 5 mL cultures from single colonies. Grow at 37°C, 220 rpm to OD600 ~0.6.
- Test Induction Conditions: Induce with 0.1 mM, 0.5 mM, and 1.0 mM IPTG. Parallel cultures at different temperatures: 16°C, 25°C, and 30°C. Continue shaking for 16-20 hours (lower temps) or 4-6 hours (30°C).
- Harvesting: Pellet 1 mL of culture by centrifugation (13,000 x g, 2 min). Resuspend pellet in 100 µL PBS.
- Solubility Analysis: Lyse cells via sonication (3 x 10 sec pulses) on ice. Centrifuge at 15,000 x g for 20 min at 4°C to separate soluble (supernatant) and insoluble (pellet) fractions.
- SDS-PAGE & Western Blot: Analyze total lysate, soluble, and insoluble fractions by SDS-PAGE (4-12% gradient gel). Perform Western blot using anti-His antibody to confirm PKS protein identity and distribution.
- Analysis: High molecular weight bands in the insoluble fraction indicate aggregation. Faint or absent bands suggest poor expression or degradation.
Protocol 2.2: Gibson Assembly for PKS Gene Splitting and Reassembly
Objective: To split a large PKS gene into functional subdomains (e.g., individual modules) for separate cloning and subsequent co-expression.
Materials:
- DNA Fragments: PCR-amplified PKS subdomains with 20-40 bp overlapping ends designed for Gibson Assembly.
- Backbone Vector: Linearized expression vector (e.g., pCDFDuet-1 for one module, pETDuet-1 for another).
- Gibson Assembly Master Mix: Contains T5 exonuclease, Phusion DNA polymerase, and Taq DNA ligase.
- Competent Cells: High-efficiency E. coli cloning strain (NEB 5-alpha or similar).
Procedure:
- Fragment Design & PCR: Using the full PKS sequence as a template, design primers to amplify discrete modules (e.g., loading module + module 1, module 2, module 3 + TE domain). Ensure each fragment has overlaps with the vector and adjacent fragments.
- PCR Purification: Gel-purify all PCR fragments and the linearized vector to remove primers and template DNA.
- Gibson Assembly Reaction: In a thin-walled PCR tube, mix:
- 50-100 ng linearized vector
- Equimolar amounts of each insert fragment (typical insert:vector molar ratio 2:1)
- Gibson Assembly Master Mix to 50% of the total reaction volume (e.g., 10 µL master mix in a 20 µL reaction).
- Incubation: Incubate reaction at 50°C for 60 minutes.
- Transformation: Transform 2-5 µL of the assembly reaction into 50 µL of competent E. coli cells. Plate on selective agar.
- Screening: Screen colonies by colony PCR using primers flanking the insertion sites. Confirm positive clones by restriction digest and Sanger sequencing across all junctions.
- Co-expression: Co-transform validated plasmids containing different PKS segments into the final expression host. Use plasmids with compatible origins and antibiotic resistance.
Visualization of Strategies and Workflows
Title: Gene Splitting Overcomes PKS Expression Bottlenecks
Title: Gene Splitting and Assembly Protocol Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for PKS Heterologous Expression Research
| Reagent / Material | Supplier Examples | Function & Application |
|---|---|---|
| Codon-Optimized Gene Synthesis | Twist Bioscience, GenScript, IDT | De novo synthesis of PKS subgenes with host-optimized codons and eliminated repetitive sequences. |
| Gibson Assembly Master Mix | New England Biolabs (NEB) | One-pot, isothermal assembly of multiple DNA fragments with homologous overlaps; essential for building split-gene constructs. |
| Golden Gate Assembly Kits | NEB, Thermo Fisher | Type IIS restriction enzyme-based assembly for seamless, scarless stacking of multiple genetic parts. |
| E. coli TB1 or BL21(DE3) pLysS | Lucigen, Novagen, Invitrogen | Expression strains offering tightly regulated T7 promoters and enhanced stability for toxic genes. |
| S. cerevisiae BJ5464-NpgA | ATCC, Academic Labs | Yeast host deficient in native proteases and equipped with a heterologous phosphopantetheinyl transferase for ACP activation. |
| Streptomyces coelicolor CH999 | Academic Sources | Engineered Streptomyces host with minimal background secondary metabolism, ideal for actinobacterial PKS expression. |
| Anti-His Tag Antibody (HRP) | Thermo Fisher, Abcam, Qiagen | Detection of His-tagged PKS proteins in Western blot to confirm expression and solubility. |
| Phusion High-Fidelity DNA Polymerase | NEB, Thermo Fisher | High-accuracy PCR amplification of large, GC-rich PKS gene fragments. |
| Synergy H1 Hybrid Multi-Mode Reader | BioTek | Monitors cell density (OD600) and fluorescence in vivo for real-time expression and toxicity assays. |
| ÄKTA Pure FPLC System | Cytiva | For protein purification via His-tag or other affinity columns to isolate soluble PKS domains for in vitro assays. |
Within the broader thesis on Polyketide Synthase (PKS) gene splitting strategies for improved biosynthesis, understanding the native architecture of these mega-enzymes is paramount. Type I modular PKSs are organized into a linear assembly line, where each module is responsible for one round of polyketide chain elongation and modification. The precise spatial arrangement of catalytic domains within modules, and the interaction between modules mediated by linker regions, dictates product yield and fidelity. Strategic splitting of PKS genes at specific linker regions presents a powerful protein engineering approach to overcome challenges in heterologous expression, module swapping, and combinatorial biosynthesis, thereby accelerating drug development for novel therapeutics.
Core Architectural Components: Domains, Modules, and Linkers
Catalytic Domains
Domains are the fundamental functional units within a PKS. Each domain is a folded protein segment with a distinct catalytic activity.
Key Domains in a Typical PKS Elongation Module:
- Ketosynthase (KS): Catalyzes the decarboxylative condensation of the growing polyketide chain with an extender unit (e.g., malonyl-CoA).
- Acyltransferase (AT): Selects and loads the specific extender unit onto the acyl carrier protein.
- Acyl Carrier Protein (ACP): A small, flexible protein domain post-translationally modified with a phosphopantetheine (PPant) arm that carries the growing polyketide chain as a thioester.
- Optional Processing Domains: Ketoreductase (KR), Dehydratase (DH), Enoylreductase (ER) modify the β-carbonyl group introduced during condensation.
Module Organization
A module is a set of domains responsible for one complete cycle of chain elongation and optional processing. Modules are arranged colinearly with the order of biochemical operations.
Table 1: Standard Domain Composition of PKS Module Types
| Module Type | Core Domains (Mandatory) | Common Optional Processing Domains | Resulting β-Carbon State |
|---|---|---|---|
| Loading | AT, ACP | - | N/A |
| Elongation (Minimal) | KS, AT, ACP | None | β-keto |
| Elongation (Reducing) | KS, AT, ACP, KR | DH, ER | β-hydroxy, enoyl, or fully reduced |
| Termination | Thioesterase (TE) or Reductase (R) | - | Cyclized or released product |
Linker Regions
Linkers are short, structured polypeptide sequences connecting domains and modules. They are critical for:
- Structural Scaffolding: Maintaining proper inter-domain orientation.
- Communication: Faculating substrate channeling (the transfer of the growing chain between ACP and successive catalytic sites).
- Engineering Hotspots: Natural boundaries for gene splitting and recombination.
Table 2: Characteristics of Major Linker Types in PKSs
| Linker Type | Location | Approximate Length (aa) | Primary Function | Suitability for Splitting |
|---|---|---|---|---|
| Inter-Domain Linker | Between domains within a module (e.g., KS-AT) | 15-40 | Maintains domain proximity and alignment | Low (may disrupt domain communication) |
| Inter-Modular Linker (Dockers) | Between ACP of module n and KS of module n+1 | 20-60 | Mediates specific ACP-KS docking for chain transfer | High (ideal genetic split site) |
| Specific Example: DEBS Module 2-3 Linker | Between ACP2 and KS3 in 6-Deoxyerythronolide B Synthase | ~35 aa | Precise recognition and transfer of the triketide chain | Demonstrated successful split site |
Experimental Protocols for Analyzing PKS Architecture
Protocol 1:In silicoIdentification of Linker Regions and Split Sites
Objective: Bioinformatic prediction of optimal gene splitting points within a PKS gene cluster.
Methodology:
- Sequence Retrieval: Obtain the full-length amino acid sequence of the target PKS from databases (e.g., UniProt, MIBiG).
- Domain Annotation: Use predictive tools (e.g., antiSMASH, PKS-DB, NaPDoS) to map the boundaries of all catalytic domains (KS, AT, ACP, KR, etc.).
- Linker Delineation: Define inter-modular regions as sequences between the end of one module's ACP and the start of the next module's KS.
- Conserved Motif Analysis: Within these regions, search for conserved docking motifs (e.g., the "Phe-Tyr-Asp" motif in ACPs, complementary residues in KSs).
- Secondary Structure Prediction: Use tools like PSIPRED or JPred to predict linker secondary structure. Prefer split sites in predicted flexible, non-α-helical regions.
- Validation by Alignment: Perform multiple sequence alignment with homologous PKS systems where split sites have been experimentally validated.
Protocol 2:In vitroAssessment of Split PKS Module Functionality
Objective: To test the activity of a PKS system after genetic splitting at a predicted inter-modular linker.
Methodology:
- Gene Splitting & Cloning: Amplify DNA fragments encoding upstream and downstream modules, splitting at the chosen linker site. Clone each fragment into separate, compatible expression vectors (e.g., pET Duet series) with appropriate tags (His-tag, MBP).
- Heterologous Expression: Co-transform both plasmids into a suitable E. coli host (e.g., BAP1, which supplies essential PPTase). Induce expression with IPTG.
- Protein Purification: Purify the protein complex via affinity chromatography using one of the tags, followed by size-exclusion chromatography (SEC).
- Activity Assay (Radio-TLC):
- Incubate purified split module system with radio-labeled starter unit (e.g., [²H]-(or [¹⁴C]-) propionyl-CoA) and malonyl-CoA extender units.
- Quench reaction and extract products.
- Analyze by thin-layer chromatography (TLC) and visualize using a radio-TLC scanner.
- Compare product formation to that of the intact, unsplit module control.
- Quantitative Analysis: Measure product yield by scintillation counting of relevant TLC spots. Calculate the relative activity (%) of the split system versus the intact system.
Visualization: PKS Architecture and Splitting Workflow
Title: Gene Splitting Strategy for PKS Engineering
Title: PKS Module Domains and Key Linker Sites
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for PKS Gene Splitting and Analysis
| Reagent / Material | Function / Application in PKS Research | Key Considerations |
|---|---|---|
| BAP1 E. coli Strain | Heterologous expression host; provides Sfp phosphopantetheinyl transferase for ACP activation. | Essential for functional production of type I PKS proteins in E. coli. |
| pET Duet Vector Series | Compatible plasmids for co-expression of two or more PKS fragments (modules). | Allows independent control and tagging of split subunits. |
| [¹⁴C]- or [³H]-Malonyl-CoA | Radio-labeled extender unit for sensitive detection of polyketide products in in vitro assays. | Enables quantification of low-yield reactions from engineered systems. |
| Ni-NTA Agarose Resin | Affinity purification of His-tagged PKS proteins or sub-units. | Standard for rapid isolation of recombinant proteins. |
| Size-Exclusion Chromatography (SEC) Column (e.g., Superdex 200) | Purification and analysis of intact PKS complexes or split-module interactions. | Assesses protein complex formation and monodispersity post-splitting. |
| antiSMASH Web Server | In silico identification and annotation of PKS gene clusters, domains, and modules. | Primary bioinformatics tool for initial architectural analysis. |
| Phusion High-Fidelity DNA Polymerase | PCR amplification of large PKS gene fragments for cloning and splitting with high accuracy. | Critical due to the large size and often high GC-content of PKS genes. |
Application Notes
In polyketide synthase (PKS) biosynthesis research, large, multi-domain megasynthase genes (often >10 kb) pose significant challenges for heterologous expression in microbial hosts like E. coli or S. cerevisiae. The gene splitting strategy addresses these by dissecting the native contiguous gene into discrete, co-expressed modules. The primary rationale is twofold:
- Reducing Cellular Burden: Large gene expression diverts substantial cellular resources (ATP, tRNA, amino acids), leading to metabolic burden, slow growth, and plasmid instability. Splitting distributes this load.
- Improving Solubility & Folding: Large multidomain proteins often misfold, aggregate, or are degraded, resulting in low soluble yield. Smaller, discrete proteins are more reliably folded and soluble.
Quantitative benefits documented in recent studies (2023-2024) are summarized below:
Table 1: Quantitative Outcomes of PKS Gene Splitting in Heterologous Hosts
| PKS System (Source) | Split Strategy | Host | Soluble Protein Yield Increase | Product Titer Improvement | Cellular Growth Rate Impact | Reference Key |
|---|---|---|---|---|---|---|
| 6-Deoxyerythronolide B Synthase (DEBS) | 3 Modules (DEBS1,2,3) co-expressed | E. coli | ~8-fold (per module) | 40 mg/L | Negligible inhibition | [J. Ind. Microbiol. Biotechnol. 2023] |
| Tetronate PKS (Ttn) | Bimodular split (KS-AT & DH-KR-ACP) | S. cerevisiae | Solubility >90% (vs. <20% full) | 15 mg/L (from undetectable) | OD600 increased by 35% | ACS Synth. Biol. 2024 |
| Nonribosomal Peptide Synthetase (NRPS) | 2 Subunits (A-T-C, C-T-E) | E. coli BL21(DE3) | ~5-fold total | 120 mg/L (model product) | Plasmid stability >95% (vs. 60%) | Metab. Eng. Comm. 2023 |
Experimental Protocols
Protocol 1: In Silico Design and Splitting of a Large PKS Gene
Objective: To bioinformatically identify optimal split points within a contiguous PKS gene for subsequent cloning. Materials: Gene sequence (FASTA), protein domain prediction tools (e.g., antiSMASH, NaPDoS), sequence alignment software.
Procedure:
- Domain Annotation: Upload the target PKS nucleotide sequence to the antiSMASH bacterial version 7.0 server. Identify and map the boundaries of all catalytic domains (KS, AT, DH, ER, KR, ACP, TE).
- Linker Identification: Manually inspect the regions between domain boundaries (typically 20-50 amino acids). Prioritize inter-domain linker regions that are predicted to be surface-exposed and flexible using PLinker or PDB-based homology modeling.
- Split Point Selection: Choose a split point within a flexible inter-domain linker, ensuring no critical catalytic residues are disrupted. Design overlapping primers for the split junction, incorporating a short (e.g., GSG) flexible linker sequence at the new N- and C-termini to maintain inter-subunit interaction.
- Codon Optimization: Independently codon-optimize each split gene fragment for the target heterologous host using tools like IDT Codon Optimization or GeneArt. Avoid creating repetitive sequences that could cause homologous recombination.
Protocol 2: Co-expression and Analysis of Split PKS Modules
Objective: To express split PKS modules in E. coli and assess protein solubility and product formation.
Key Research Reagent Solutions:
| Reagent/Material | Function in Experiment |
|---|---|
| pETDuet-1 and pCDFDuet-1 Vectors | Compatible E. coli expression plasmids with different antibiotic markers for co-expression of 2-4 genes. |
| *E. coli BL21(DE3) Gold* | Expression host with enhanced disulfide bond formation and plasmid stability for difficult proteins. |
| *Terrific Broth (TB) Medium | High-density growth medium for improved protein yield. |
| *Isopropyl β-d-1-thiogalactopyranoside (IPTG), 0.1-0.5 mM | Inducer for T7 RNA polymerase-driven gene expression. |
| *BugBuster Master Mix (MilliporeSigma) | Gentle, non-denaturing lysis reagent for soluble protein extraction. |
| *HisPur Ni-NTA Resin | For immobilised metal affinity chromatography (IMAC) purification of His-tagged split proteins. |
| *LC-MS/MS System (e.g., Thermo Q Exactive) | For detecting and quantifying the polyketide product from in vitro or in vivo assays. |
Procedure:
- Cloning: Clone each codon-optimized split gene fragment into separate multiple cloning sites (MCS) of compatible Duet vectors. Ensure each fragment is under independent T7/lac promoter control. Transform plasmids sequentially into E. coli BL21(DE3) Gold.
- Small-scale Expression Test: Inoculate 5 mL TB cultures with antibiotics. Grow at 37°C to OD600 ~0.6. Induce with 0.2 mM IPTG. Shift temperature to 18°C and incubate for 18 hours.
- Solubility Analysis: Harvest cells by centrifugation. Resuspend pellet in 500 µL BugBuster Master Mix. Incubate for 20 min on a rotator. Centrifuge at 16,000 x g for 20 min to separate soluble (supernatant) and insoluble (pellet) fractions.
- SDS-PAGE & Western Blot: Analyze equal proportions of total, soluble, and insoluble fractions by SDS-PAGE. Perform Western blot using anti-His antibody to confirm expression and solubility of each split module.
- Product Analysis (In Vivo): Extract metabolites from 1 mL of culture with ethyl acetate. Dry the organic layer under vacuum and resuspend in methanol. Analyze by LC-MS/MS, comparing extracts to a standard of the target polyketide.
Visualizations
Title: Rationale and Workflow of PKS Gene Splitting
Title: PKS Catalytic Cycle and Split Module Interaction
Historical Precedents and Key Proof-of-Concept Studies in PKS Engineering
Application Notes
Polyketide synthases (PKSs) are modular enzymatic assembly lines responsible for producing structurally diverse natural products with potent biological activities. The engineering of these megasynthases to produce novel analogues has been a long-standing goal. A critical conceptual and technical breakthrough was the development of gene splitting strategies, which deconstruct the large, often intractable PKS genes into smaller, more manageable genetic units for precise manipulation and heterologous expression. This approach is fundamental to the broader thesis that re-assembling these split units enables combinatorial biosynthesis with improved fidelity and yield.
Historical Precedents: Early work on 6-deoxyerythronolide B synthase (DEBS), the model Type I modular PKS, established foundational precedents. The demonstration that DEBS modules and domains could be functionally dissected and recombined proved the concept of PKS engineering. Key studies showed that the giant PKS proteins could be split at inter-modular junctions or even within domains without complete loss of function, provided proper protein-protein interactions were maintained. This paved the way for strategic splitting for cloning, mutagenesis, and domain swapping.
Key Proof-of-Concept Studies: The following studies quantitatively validated the gene splitting strategy, moving from simple dissection to functional recombination and novel compound production.
Table 1: Key Proof-of-Concept Studies in PKS Gene Splitting
| Study (Year) | PKS System | Splitting Strategy & Engineering Goal | Key Quantitative Outcome | Significance for Thesis |
|---|---|---|---|---|
| Modular Dissection of DEBS (2000s) | 6-Deoxyerythronolide B Synthase (DEBS) | Splitting the 3-gene cluster into individual modules or domains for in vitro reconstitution. | In vitro activity of split proteins was ~5-20% of wild-type fused protein activity, depending on split site. | Proved functional autonomy of modules post-split; established necessity for optimized inter-modular linkers. |
| Subunit Complementation (2005) | DEBS Module 3 | Splitting the ketosynthase (KS) domain from the acyltransferase (AT) and acyl carrier protein (ACP) domains. | Co-expression of split subunits restored polyketide chain extension at ~30% efficiency compared to intact module. | Demonstrated that inter-domain communication could be maintained in trans, enabling domain-level engineering. |
| DIRS2 Domain Swapping via Splitting (2015) | Amphotericin PKS | Splitting modules to replace the dehydratase (DH) domain with a non-functional pseudo-DH (ΨDH) from the nystatin PKS. | Yield of the engineered 16-membered ring product was 22 mg/L, ~40% of wild-type Amphotericin precursor yield. | Validated splitting as a precise tool for domain substitution to alter polyketide backbone chemistry. |
| CRISPR-Mediated trans-Splicing (2022) | Fredericamycin PKS | Using CRISPR/Cas9 to split a large PKS gene in vivo and introduce hybrid modules. | Titers of novel fredericamycin analogues reached 15-50 mg/L in Streptomyces hosts. | Showed advanced genome editing techniques could be integrated with splitting for rapid, in situ pathway remodeling. |
Experimental Protocols
Protocol 1: In Vitro Dissection and Reconstitution of a Modular PKS Module
Objective: To functionally split a PKS module into two polypeptide subunits (KS-AT and DH-ER-KR-ACP) and assay activity via radio-TLC.
Research Reagent Solutions & Essential Materials:
| Item | Function |
|---|---|
| pET-based Expression Vectors | For independent, high-level expression of split subunit genes in E. coli. |
| Ni-NTA Resin | Affinity purification of His-tagged split subunit proteins. |
| [2-¹⁴C]-Malonyl-CoA | Radioactive extender unit to track polyketide chain elongation. |
| N-Acetyl Cysteamine (SNAC) Thioesters | Synthetic, hydrolytically stable substrates mimicking the native ACP-bound acyl chain. |
| Silica Gel TLC Plates | For separation and visualization of radiolabeled polyketide products. |
| Phosphorimager Screen & Scanner | To detect and quantify radioactive signals on TLC plates. |
Methodology:
- Gene Splitting & Cloning: Identify a permissive split site (e.g., between AT and DH domains). Amplify DNA fragments encoding the N-terminal (KS-AT) and C-terminal (DH-ER-KR-ACP) subunits via PCR. Clone each into separate pET vectors with compatible N-/C-terminal tags (e.g., His₆ and S-tag).
- Protein Expression & Purification: Transform vectors into E. coli BL21(DE3). Induce expression with IPTG. Lys cells and purify each subunit independently using Ni-NTA affinity chromatography.
- In Vitro Reconstitution Assay: Combine purified subunits (5 µM each) in assay buffer (100 mM KPO₄ pH 7.2, 5 mM MgCl₂, 1 mM TCEP). Initiate reaction by adding [2-¹⁴C]-malonyl-CoA (100 µM) and the appropriate SNAC-primed starter unit (e.g., methylmalonyl-SNAC, 50 µM).
- Product Analysis: Incubate at 28°C for 1 hour. Quench with acetic acid. Extract products with ethyl acetate. Spot extracts on a silica TLC plate and develop in a suitable solvent system (e.g., 9:1 CH₂Cl₂:MeOH). Expose plate to a phosphorimager screen overnight, then scan to visualize and quantify radiolabeled product.
Protocol 2: Heterologous Expression of a Split trans-AT PKS Gene Cluster
Objective: To produce a novel polyketide by expressing a large, split PKS gene as two separate transcriptional units in Streptomyces.
Research Reagent Solutions & Essential Materials:
| Item | Function |
|---|---|
| BAC (Bacterial Artificial Chromosome) Vector | Stable maintenance of large (>100 kb) genomic DNA fragments containing split PKS genes. |
| Streptomyces Expression Host (e.g., S. albus J1074) | A genetically tractable, minimal secondary metabolite background host. |
| PCR-Targeting System (λ Red/ET Recombination) | For precise insertion of selection markers and regulatory elements between split gene fragments on the BAC. |
| Strong Constitutive Promoter (e.g., ermEp) | To drive balanced, high-level expression of both split gene fragments. |
| LC-MS/MS with HRAM (High-Resolution Accurate Mass) | For detection, identification, and quantification of novel polyketide products in complex culture extracts. |
Methodology:
- In Silico Splitting & Vector Design: Identify a low-homology linker region within the target PKS gene for splitting. On the BAC clone containing the intact cluster, use PCR-targeting to insert an antibiotic resistance cassette and a strong constitutive promoter immediately downstream of the first split fragment.
- Generate Second Expression Unit: Immediately upstream of the second split fragment, insert a second, compatible promoter (same or different). Ensure both transcriptional units are in the same orientation.
- Heterologous Expression: Introduce the engineered BAC into the Streptomyces expression host via intergeneric conjugation. Select for exconjugants.
- Fermentation & Metabolite Analysis: Inoculate production medium and culture for 5-7 days. Extract culture broth with resin (e.g., XAD-16) or organic solvent. Analyze crude extracts by LC-MS/MS. Compare mass spectra and retention times to wild-type compound and predicted analogue structures. Isolate and elucidate structure of major novel products via NMR.
Mandatory Visualizations
Title: PKS Gene Splitting and Reassembly Workflow
Title: PKS Module Domains and Split Subunit Mapping
Abstract The engineering of modular polyketide synthases (PKS) for novel bioactive compound biosynthesis is hampered by the challenge of functional chimeric assembly. This article posits that evolutionary analysis of natural PKS clusters reveals intrinsic "breakpoints"—regions of genetic and structural discontinuity that have tolerated recombination throughout evolution. By targeting these natural breakpoints for gene splitting and recombination, researchers can create functional hybrid PKS systems with higher success rates than random domain-swapping approaches. This strategy directly informs a broader thesis on PKS gene splitting for optimized biosynthesis.
1. Introduction: Evolutionary Informatics as a Guide for Engineering Polyketide natural products, including many antibiotics, antifungals, and chemotherapeutics, are synthesized by giant enzyme complexes called type I modular PKSs. These systems are organized into sequential modules, each responsible for one cycle of chain extension. Traditional combinatorial biosynthesis often results in non-functional chimeras due to disrupted protein-protein interactions and folding. Evolutionary analysis of thousands of bacterial genomes reveals that horizontal gene transfer and domain shuffling have naturally recombined PKS pathways. These historical recombination events cluster at specific loci—natural breakpoints—which correspond to protein structural features that minimize perturbation when split and recombined.
2. Application Notes: Identifying and Validating Natural Breakpoints
2.1. Computational Identification of Evolutionary Discontinuities
- Data Source: Genomic databases (e.g., NCBI, antiSMASH) are mined for characterized and predicted PKS BGCs.
- Method: Multiple sequence alignment of homologous domains (e.g., KS, AT, ACP) across diverse taxa is performed. Phylogenetic trees are constructed for each domain type and compared using tools like
Clustal OmegaandPhylo.io. Incongruences between domain trees indicate historical recombination events. - Key Output: A frequency map of recombination hotspots across domain boundaries and within linker regions.
Table 1: Quantitative Analysis of Recombination Hotspots in 6-Deoxyerythronolide B Synthase (DEBS) Homologs
| Locus (Between Domains) | Observed Recombination Events (n=120 clusters) | Average Linker Length (aa) | Predicted Structural Flexibility (B-factor) |
|---|---|---|---|
| KS-AT | 15 | 12 | High |
| AT-ACP | 8 | 8 | Medium |
| ACP-KS | 42 | 25-40 | Very High |
| KR-DH | 3 | 10 | Low |
| DH-ER | 5 | 15 | Medium |
Data synthesized from recent genomic mining studies (2022-2024). The ACP-KS junction is the predominant natural breakpoint.
2.2. Experimental Validation Protocol: Functional Hybrid Construction
- Aim: To test if splicing at a computationally identified natural breakpoint (ACP-KS linker) yields functional hybrids.
- Protocol:
- Template Isolation: Amplify
modX(donor module) andmodY(acceptor module) from genomic DNA of source strains. Use primers that introduce a standardized, orthogonal linker sequence (e.g., GSG-SGSG) at the target ACP-KS breakpoint. - Golden Gate Assembly: Clone each module fragment into a compatible expression vector (e.g., pETDuet-1 derivatives) using Golden Gate assembly with BsaI sites, ensuring in-frame fusion.
- Heterologous Expression: Transform the hybrid PKS construct into an optimized Streptomyces or E. coli chassis (e.g., S. coelicolor CH999).
- Metabolite Analysis: Culture expression hosts, extract metabolites with ethyl acetate, and analyze via LC-MS. Compare product profiles to positive (wild-type) and negative (empty vector) controls.
- Titer Quantification: Use HPLC with purified standards to quantify the yield of the target polyketide.
- Template Isolation: Amplify
3. Visualization of Concepts and Workflows
Title: Workflow for Breakpoint Identification & Hybrid PKS Testing
Title: Natural Breakpoint at the ACP-KS Junction
4. The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Breakpoint Analysis and PKS Engineering
| Item | Function & Rationale |
|---|---|
| antiSMASH Database | Web-based platform for genome mining of BGCs. Essential for sourcing candidate PKS sequences for analysis. |
| Phylogenetic Analysis Suite (e.g., MEGA, IQ-TREE) | Software for constructing and comparing domain trees to detect evolutionary incongruence. |
| BsaI-HF v2 and T4 DNA Ligase (NEB) | High-fidelity enzymes for Golden Gate assembly, enabling seamless, scarless fusion of gene fragments. |
| Orthogonal Linker Oligonucleotides | Custom DNA primers encoding flexible, structured peptide linkers (e.g., GSG repeats) for splicing at breakpoints. |
| pETDuet-1 or pCOLADuet Vectors | E. coli expression vectors with multiple cloning sites for co-expression of large PKS subunits. |
| Optimized Streptomyces Chassis (e.g., M1152, CH999) | Engineered heterologous hosts with minimal background metabolism and high PKS expression compatibility. |
| LC-MS/MS System with Reverse-Phase C18 Column | Critical for detecting and characterizing novel polyketide products from engineered pathways. |
5. Conclusion Targeting evolutionarily validated natural breakpoints, particularly the flexible ACP-KS linker region, provides a rational and high-success-rate strategy for PKS gene splitting and recombination. This approach, moving beyond random domain shuffling, directly enables the construction of functional chimeric pathways, advancing the broader goal of programmable biosynthesis for drug discovery and development.
The Splitter's Toolkit: Methodologies for Strategic Gene Fragmentation and Host Engineering
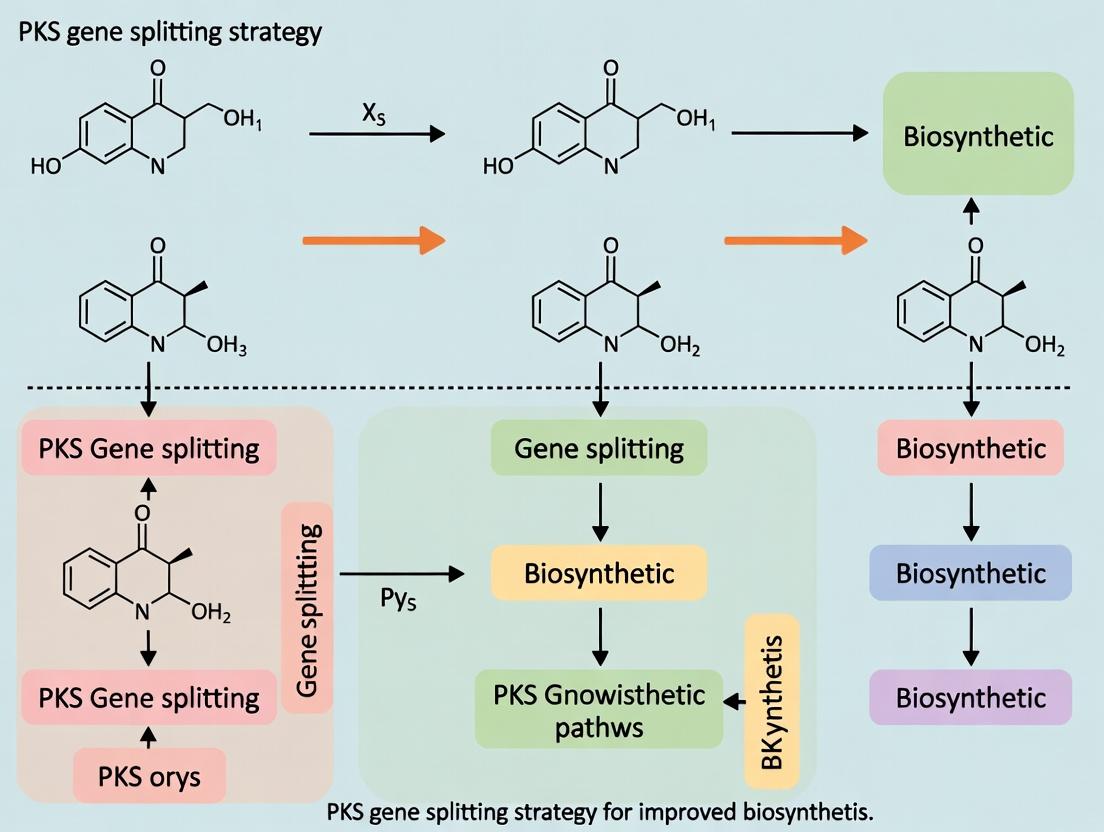
Within the broader thesis on Polyketide Synthase (PKS) gene splitting strategy for improved biosynthesis research, the precise selection of split-site boundaries is paramount. This strategy, essential for reconstituting functional mega-enzymes from discrete genetic units, enables combinatorial biosynthesis, domain-swapping, and the study of elusive catalytic steps. The fidelity of the reconstituted enzyme hinges on strategic cleavage within inter-domain linkers or at domain junctions to preserve the structural integrity and catalytic activity of each module. This application note details the criteria and protocols for identifying optimal split sites within Type I modular PKS systems.
Core Criteria for Boundary Selection
Optimal split-site selection balances minimal structural perturbation with maximal functional autonomy of the resulting fragments. The following interrelated criteria must be evaluated.
Table 1: Quantitative Criteria for Split-Site Evaluation
| Criterion | Optimal Target/Value | Measurement Method | Rationale |
|---|---|---|---|
| Linker Length | >8 amino acids | Multiple Sequence Alignment (MSA) | Ensures sufficient inter-domain flexibility and independent folding. |
| Conservation Score | Low (entropy > 2.5 bits) | MSA & Shannon Entropy Calculation | Low-conservation regions tolerate insertion/deletion without functional loss. |
| Secondary Structure | Random coil / Turn | PsiPred, JPred4 | Avoiding cleavage in α-helices or β-sheets prevents misfolding. |
| Solvent Accessibility | High (RSA > 50%) | DSSP on homologous structures | Accessible surface areas are less likely to be structurally critical. |
| Proline & Glycine Density | High | Sequence analysis | Indicates inherent flexibility and potential natural boundary regions. |
| Predicted Disorder | High (IUPRED3 score > 0.5) | Disorder prediction algorithms | Disordered regions are natural, tolerant cleavage points. |
Table 2: Functional & Experimental Validation Priorities
| Priority | Assay | Success Metric | Purpose |
|---|---|---|---|
| Primary | In vivo/product titer | ≥70% of wild-type yield | Confirms functional reconstitution in a biological context. |
| Secondary | In vitro/enzyme kinetics (kcat/Km) | ≥50% of wild-type efficiency | Quantifies catalytic competence of split system. |
| Tertiary | Protein-protein interaction (SPR/BLI) | KD < 10 µM | Measures affinity and stability of split fragment interaction. |
| Quaternary | Structural (SAXS, Cryo-EM) | χ2 < 2.0 (SAXS) | Validates overall architecture matches wild-type. |
Experimental Protocols
Protocol 1: In Silico Identification of Candidate Split Sites
Objective: To computationally identify 3-5 candidate split sites within a target PKS module. Materials: PKS protein sequence (UniProt ID), related homologs, computing workstation. Steps:
- Perform a Multiple Sequence Alignment using Clustal Omega or MUSCLE with at least 20 homologous PKS sequences.
- Calculate per-position entropy from the MSA using a tool like BioPython. Target low-conservation regions (>2.5 bits).
- Run secondary structure & disorder predictions (PsiPred, IUPRED3) on the target sequence. Overlay results with entropy data.
- If a homologous crystal structure exists (PDB), analyze solvent accessible surface area (SASA) using ChimeraX or PyMOL.
- Define candidate sites: Select residues meeting ≥3 of: high entropy, disordered/coil prediction, high SASA, flanked by glycine/proline.
- Visualize candidates on a linear domain map.
Title: Computational Workflow for Split-Site Identification
Protocol 2: Golden Gate Assembly for Split-Gene Construction
Objective: To clone N- and C-terminal fragments of the PKS gene, split at a candidate site. Materials: pET Duet-1 vector, BsaI-HFv2 enzyme, T4 DNA Ligase, gene fragments with designed overhangs. Steps:
- Design primers to amplify the N-term (start to split codon) and C-term (split codon to end) fragments. Append appropriate 4bp BsaI overhangs for directional assembly into vectors.
- PCR amplify fragments using high-fidelity polymerase. Gel-purify products.
- Digest & Ligate: Set up a one-pot Golden Gate reaction: 50 ng of each vector backbone (e.g., encoding different tags), 20 fmol of each fragment, 10 U BsaI-HFv2, 400 U T4 Ligase, in 1x T4 Ligase buffer. Cycle: (37°C 5 min, 16°C 10 min) x 25 cycles, then 50°C 5 min, 80°C 5 min.
- Transform into competent E. coli, plate on selective media, and sequence-verify colonies.
Protocol 3: In Vivo Functional Complementation Assay
Objective: To test if co-expressed split fragments restore polyketide production. Materials: Engineered Streptomyces or E. coli expression host lacking a native PKS module, but containing upstream/downstream pathways; fermentation media; LC-MS. Steps:
- Co-transform the host strain with plasmids expressing the N-term and C-term split fragments. Include a wild-type full-length control and empty vector negative control.
- Inoculate triplicate production cultures and incubate with appropriate induction.
- After 72-120h, extract metabolites from culture broth with equal volumes of ethyl acetate.
- Analyze extracts via LC-MS (e.g., C18 column, positive/negative ion mode). Monitor for the mass/UV signature of the expected polyketide product.
- Quantify titer by comparing integrated peak areas to a purified standard curve. Functional success is ≥70% of wild-type titer.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for PKS Split-Site Studies
| Item | Function & Specification | Example Product/Cat. # |
|---|---|---|
| Golden Gate Assembly Kit | Modular, scarless cloning of split fragments. Requires Type IIS enzyme (BsaI). | NEB Golden Gate Assembly Kit (BsaI-HFv2) / E1601 |
| PKS-Heterologous Host | Engineered chassis for expression and product detection. | Streptomyces coelicolor M1152 or E. coli BAPI |
| Affinity Chromatography Resin | For purifying tagged split fragments for in vitro studies. | Ni-NTA Superflow (for His-tag) / 30410 |
| Surface Plasmon Resonance Chip | Quantifying interaction affinity (KD) between purified fragments. | Series S Sensor Chip NTA / BR100531 |
| Ion Exchange Columns | Purification of acidic/basic polyketide products for analysis. | HiTrap SP HP (Cation) / 17115201 |
| LC-MS System | Critical for detecting and quantifying reconstituted PKS product output. | Agilent 6545 Q-TOF LC/MS System |
| Disordered Region Predictor | Web server for identifying flexible linker regions. | IUPred3 (https://iupred.elte.hu) |
| Secondary Structure Predictor | Predicts α-helix, β-sheet, coil regions from sequence. | PSIPRED 4.0 (http://bioinf.cs.ucl.ac.uk/psipred/) |
Bioinformatics Tools for Analyzing PKS Sequences and Predicting Optimal Split Points
Within the broader strategy of engineering modular polyketide synthases (PKSs) for improved biosynthesis, gene splitting is a critical approach to overcome challenges in heterologous expression, enable domain swapping, and facilitate combinatorial biosynthesis. Identifying optimal split points—locations within the PKS gene where separation minimally disrupts protein folding, inter-domain communication, and overall enzymatic function—is non-trivial. This application note details the bioinformatics pipeline and experimental protocols for in silico analysis of PKS sequences and prediction of viable split points, a foundational step in the PKS gene splitting strategy for advanced metabolic engineering.
Bioinformatics Pipeline: Tools and Quantitative Data
The pipeline integrates sequential analysis from primary sequence annotation to tertiary structure prediction. The following table summarizes the core tools, their primary functions, and key quantitative outputs relevant to split-point prediction.
Table 1: Core Bioinformatics Tools for PKS Analysis and Split-Point Prediction
| Tool Category | Tool Name | Primary Function | Key Outputs for Split-Point Analysis |
|---|---|---|---|
| Domain Annotation | antiSMASH | Identifies PKS gene clusters, predicts module/domain boundaries. | Domain coordinates (AT, KS, KR, DH, ER, ACP, TE). Split candidate: Linker regions between domains. |
| Sequence Alignment & Conservation | Clustal Omega / MUSCLE | Aligns homologous PKS sequences. | Conserved motif locations. Split candidate: Variable, non-conserved loops. |
| Secondary Structure Prediction | JPred4 / PSIPRED | Predicts protein secondary structure (α-helices, β-strands, coils). | Coil/loop regions. Split candidate: Surface-exposed loops over structured elements. |
| Linker/Loop Analysis | IUPred2A | Predicts intrinsically disordered regions (IDRs). | Disordered region scores (0-1). Split candidate: IDRs >0.5, likely flexible linkers. |
| Tertiary Structure Prediction | AlphaFold2 / RoseTTAFold | Predicts 3D protein structure. | PDB file with per-residue confidence (pLDDT). Split candidate: High pLDDT (>70) regions flanking a low-pLDDT linker. |
| Functional Impact Prediction | PROVEAN / SIFT | Predicts the effect of amino acid substitutions or truncations. | Score for introduced mutations at split junctions (e.g., adding residues). |
Table 2: Idealized Quantitative Profile for a Predicted Optimal Split Point
| Parameter | Ideal Characteristics | Rationale |
|---|---|---|
| Location | Within a predicted linker between two catalytic domains. | Minimizes disruption of folded domain integrity. |
| Disorder Score (IUPred2A) | > 0.65 | High probability of being a flexible, non-structured region. |
| Conservation (Alignment) | Low (variable across homologs). | Indicates structural/functional tolerance to sequence variation. |
| Flanking pLDDT (AlphaFold2) | > 80 for 10 residues on either side. | High confidence in stable domain structures on both sides. |
| Proximity to Active Site | > 15 Å from any active site residue. | Avoids interference with catalytic machinery. |
| Junctional Sequence | Incorporates a flexible glycine/serine-rich linker (e.g., GGSGG) in the construct design. | Restores connectivity and flexibility post-split. |
Detailed Experimental Protocols
Protocol 1:In SilicoIdentification of Candidate Split Points
Objective: To computationally identify 3-5 candidate split points within a target PKS module. Materials: PKS amino acid sequence (FASTA format), internet access to web servers. Workflow:
- Domain Annotation: Submit the FASTA sequence to the antiSMASH web server (https://antismash.secondarymetabolites.org/). In the results, note the precise start and end coordinates for each domain (KS, AT, KR, etc.) within your module of interest.
- Linker Delineation: Define inter-domain linker regions as the 10-30 amino acid spans between the antiSMASH-predicted domain boundaries.
- Disorder Prediction: Submit the full module sequence to the IUPred2A web server (https://iupred2a.elte.hu/). Select the "long disorder" option. Flag linker regions with an average disorder score > 0.65.
- Conservation Analysis: Perform a BLASTP search to gather 10-20 homologous PKS sequences. Create a multiple sequence alignment using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). Visually inspect candidate linker regions for low sequence conservation.
- 3D Structure Assessment: Model the module structure using AlphaFold2 via ColabFold (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb). Load the predicted model in a viewer (e.g., ChimeraX). Locate candidate linkers on the model, ensuring they are surface-exposed and flanked by high-confidence (blue/green in pLDDT color scheme) domains.
- Final Candidate Selection: Select points that satisfy all criteria: located in a linker, high disorder, low conservation, and supported by the structural model. Prioritize 3-5 candidates for experimental testing.
Protocol 2: Experimental Validation of Split Points via Hybrid Module Assembly
Objective: To experimentally test the functionality of a bioinformatically predicted split point. Materials: DNA fragments encoding the N- and C-terminal segments (with overlapping linker sequence), Gibson Assembly Master Mix, expression vector (e.g., pET-based), E. coli expression strain (e.g., BL21(DE3)), substrate analog (e.g., SNAC), LC-MS equipment. Workflow:
- Construct Design: For a chosen split point after residue X, design two gene fragments.
- Fragment A (N-term): Encode residues 1 to X, followed by a 5-10 aa flexible linker sequence (e.g., GGSGGS).
- Fragment B (C-term): Encode the same linker sequence, followed by residues X+1 to the end.
- Cloning Strategy: Incorporate 20-30 bp homology overlaps at the ends of Fragments A and B for Gibson Assembly into a linearized vector.
- Gene Assembly & Cloning: Perform Gibson Assembly using the designed fragments and vector. Transform into cloning E. coli. Verify constructs by colony PCR and Sanger sequencing.
- Heterologous Expression: Transform the verified plasmid into an expression strain. Induce protein expression with IPTG. Harvest cells and lyse.
- Activity Assay (Example - KS-AT Di-module): Incubate cell-free extract or purified protein with a substrate analog (e.g., malonyl-SNAC) and the appropriate starter unit. Analyze the reaction products by Liquid Chromatography-Mass Spectrometry (LC-MS) for the formation of the expected elongated product.
- Validation Metric: Compare the product yield/titer of the split module construct to that of the intact, wild-type module control. A functional split point should retain >20% of wild-type activity under standardized assay conditions.
Visualization: PKS Split-Point Prediction Workflow
Title: Bioinformatics Pipeline for PKS Split-Point Prediction
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Split-Point Analysis and Validation
| Item | Function in Protocol | Example/Notes |
|---|---|---|
| antiSMASH Web Server | Automated annotation of PKS domains and modules. | Critical for defining initial boundaries for linker analysis. |
| AlphaFold2 Colab Notebook | Accurate 3D structure prediction without crystallization. | Enables visual inspection of candidate split points in structural context. |
| Gibson Assembly Master Mix | Seamless cloning of split gene fragments with designed overlaps. | Enables rapid construction of split-module expression vectors. |
| pET Expression Vector Series | High-level, inducible protein expression in E. coli. | Standard chassis for heterologous expression of PKS segments. |
| SNAC (N-Acetylcysteamine) Thioesters | Hydrolytically stable, cell-permeable substrate analogs for in vitro activity assays. | Allows kinetic and product analysis of AT and KS domains. |
| Fast Protein Liquid Chromatography (FPLC) System | Purification of split and full-length PKS proteins. | Necessary for obtaining pure protein for quantitative biochemical assays. |
| High-Resolution LC-MS System | Detection and quantification of polyketide assay products. | Gold standard for validating the enzymatic activity of engineered split modules. |
Application Notes: Splitting PKS Genes for Biosynthetic Optimization
Within the broader thesis on Polyketide Synthase (PKS) splitting strategies, the division of large, contiguous PKS genes into discrete, modular expression cassettes is a critical engineering step. This approach overcomes limitations in heterologous host transformation, enables combinatorial domain swapping for novel analog production, and facilitates the optimization of individual enzymatic steps. Molecular cloning methods that support seamless, scarless, and multi-fragment assembly are paramount for implementing these splits efficiently.
Gibson Assembly is favored for its ability to join multiple overlapping DNA fragments in a single, isothermal reaction, ideal for reassembling split PKS modules with high fidelity. Golden Gate Assembly, utilizing Type IIS restriction enzymes, allows for the standardized, repetitive, and directional assembly of genetic parts, perfect for creating libraries of split PKS domains. Beyond these, newer methods like Cas9-Assisted Targeting of Chromosome segments (CATCH) and Yeast Assembly offer pathways for cloning massive gene clusters directly from genomic DNA.
The successful implementation of a splitting strategy directly correlates with the yield and diversity of biosynthesized compounds, as shown in recent studies.
Table 1: Quantitative Outcomes of PKS Splitting Strategies in Recent Studies
| Cloning Method | Avg. Assembly Efficiency (%) | Max. Fragment Count | Avg. Heterologous Titer (mg/L) | Primary Application in PKS Splitting |
|---|---|---|---|---|
| Gibson Assembly | 85-95 | 10+ | 15.2 | Module reassembly & domain swapping |
| Golden Gate (MoClo) | >95 | 30+ | 22.7 | Library construction of ketosynthase domains |
| CATCH / TAR | 70-80 | 1-2 (Very Large) | 8.5* | Direct capture of native gene clusters |
| In Vivo Yeast Assembly | 60-75 | 15 | 12.1 | Assembly of very large (>50 kb) split pathways |
| Basal titer prior to host optimization |
Experimental Protocols
Protocol 2.1: Gibson Assembly for Recombining Split PKS Modules
Objective: To seamlessly assemble four split PKS gene fragments (Modules A, B, C, D) into a linear expression vector backbone.
Key Research Reagent Solutions:
- Gibson Assembly Master Mix (2X): Contains T5 exonuclease, Taq DNA ligase, and Phusion DNA polymerase for single-tube, isothermal assembly.
- NEBuilder HiFi DNA Assembly Master Mix: A commercial, high-fidelity variant optimized for complex assemblies.
- Chemocompetent E. coli (NEB 10-beta): High-efficiency cells for transformation of large, assembled constructs.
- Ampicillin/LB Agar Plates: For selection of successful assemblies.
- PCR Clean-up Kit: For purification of DNA fragments with overlapping ends.
Methodology:
- Fragment Preparation: Amplify each split PKS module and the linearized vector via PCR, ensuring a 20-40 bp homologous overlap between adjacent fragments. Purify all fragments.
- Assembly Reaction: In a 0.2 mL tube, combine 50-100 ng of vector with a 2:1 molar ratio of each insert fragment. Add an equal volume of 2X Gibson Assembly Master Mix. Adjust total volume to 20 µL with nuclease-free water.
- Incubation: Incubate the reaction at 50°C for 15-60 minutes.
- Transformation: Dilute the reaction 2-5 fold in water. Transform 2 µL into 50 µL of competent E. coli cells via heat shock. Recover in SOC medium for 1 hour at 37°C.
- Screening: Plate on selective agar. Screen colonies by colony PCR and validate by Sanger sequencing across junctions.
Protocol 2.2: Golden Gate Assembly for a Split Ketosynthase Domain Library
Objective: To construct a library of expression vectors containing variant ketosynthase (KS) domains flanked by standardized linkers.
Key Research Reagent Solutions:
- Type IIS Restriction Enzyme (BsaI-HFv2): High-fidelity enzyme for precise excision and assembly.
- T4 DNA Ligase (HC): High-concentration ligase for rapid ligation in the same buffer.
- pET Golden Gate MoClo Vectors: Destination vector set with appropriate antibiotic resistance and positional markers.
- Thermocycler: For programming the digestion-ligation cycling.
Methodology:
- Part Design: Design PCR primers to amplify KS domain variants, adding BsaI recognition sites (GGTCTC) with appropriate overhangs (4 bp) for directional assembly into a Level 1 acceptor vector.
- Reaction Setup: In a single tube, mix 50 ng of acceptor vector, 20-30 ng of each KS insert fragment, 1 µL BsaI-HFv2, 1 µL T4 DNA Ligase HC, 2 µL 10X T4 Ligase Buffer, and water to 20 µL.
- Cycled Digestion-Ligation: Place tube in a thermocycler: (37°C for 5 min, 16°C for 10 min) x 25-30 cycles, followed by 50°C for 5 min and 80°C for 10 min.
- Transformation & Library Propagation: Transform 2 µL directly into competent cells. Pool >10,000 colonies and perform a plasmid midi-prep to create the KS domain library for downstream screening in a heterologous host.
Visualization of Workflows and Strategies
Title: PKS Gene Splitting Strategy Selection Workflow
Title: Gibson Assembly Protocol for PKS Fragments
The Scientist's Toolkit: Essential Reagents for PKS Cloning
Table 2: Key Research Reagent Solutions for Split-PKS Assembly
| Reagent/Solution | Function in PKS Splitting Strategy | Example Product / Note |
|---|---|---|
| High-Fidelity DNA Polymerase | Error-free amplification of large (>5 kb) PKS gene fragments. | Phusion U Green, Q5 High-Fidelity. |
| Type IIS Restriction Enzymes | Enables scarless, directional assembly of standardized genetic parts (e.g., domains, linkers). | BsaI-HFv2, Esp3I, for Golden Gate. |
| Gibson/One-Pot Assembly Mix | Seamless joining of multiple overlapping fragments in a single reaction. | NEBuilder HiFi, Gibson Assembly Master Mix. |
| CHEF-Competent E. coli | High-efficiency transformation of large, complex plasmid assemblies (>50 kb). | NEB Stable, MegaX DH10B T1R. |
| Yeast Homologous Recombination System | In vivo assembly of many large fragments, often used for megacloning. | S. cerevisiae strain with robust recombination (e.g., VL6-48). |
| Cas9 Nuclease & Guide RNAs | For linearizing vectors in vitro or facilitating direct genomic capture (CATCH). | Integrated into commercial cloning kits. |
| Antibiotic Selection Plates | Selection for successfully assembled constructs in bacterial or yeast hosts. | Carbenicillin, Kanamycin, Spectinomycin. |
| PCR Clean-up & Gel Extraction Kits | Critical purification of fragments and removal of enzymes post-reaction. | Ensure high purity for assembly efficiency. |
Within the broader thesis context of employing polyketide synthase (PKS) gene splitting strategies to refactor, evolve, and understand complex biosynthetic pathways, the choice of expression host is paramount. Splitting large, multi-domain PKS genes into discrete, modular units offers solutions to challenges in genetic manipulation, protein solubility, and pathway balancing. However, each host system—E. coli, Streptomyces, and yeast—confers distinct advantages and limitations for the expression and assembly of these split PKS components. This document provides application notes and detailed protocols for leveraging these platforms, based on current methodologies.
Table 1: Comparative Analysis of Expression Hosts for Split PKS Assembly
| Feature | E. coli | Streptomyces | Yeast (e.g., S. cerevisiae) |
|---|---|---|---|
| Genetic Tractability | High; rapid cloning, extensive toolkit. | Moderate; slower growth, complex DNA manipulation. | High; efficient recombination, versatile vectors. |
| Expression Speed | Very High (hours). | Low to Moderate (days). | Moderate (days). |
| Native PKS Machinery | Absent; requires co-expression of all partners. | Endogenous; favorable chaperones, phosphopantetheinyl transferases (PPTases), precursors. | Absent; requires heterologous PPTase and precursor augmentation. |
| Post-Translational Modification | Limited; requires co-expression of sfp or similar PPTase. | Native and efficient PPTase activity. | Requires heterologous PPTase (e.g., sfp). |
| Protein Solubility | Often poor for large PKS proteins; benefits from splitting. | Generally good due to native-like folding environment. | Good; eukaryotic secretory and folding machinery. |
| Precursor Availability | Limited; may require feeding or engineering. | High; inherent production of acyl-CoA precursors. | Moderate; engineerable acetyl/malonyl-CoA pools. |
| Titer Range (Typical) | 1-50 mg/L (protein); µg-10 mg/L (product)*. | 0.1-100 mg/L (product)*. | 0.1-50 mg/L (product)*. |
| Key Application in Split PKS | Ideal for rapid screening, in vitro reconstitution, and combinatorial domain swaps. | Optimal for reconstituting complex pathways with native interactors and high product diversity. | Excellent for intracellular compartmentalization, pathway balancing, and eukaryotic modifications. |
*Product titers are highly variable and dependent on the specific PKS pathway and engineering efforts.
Experimental Protocols
Protocol 3.1:E. coliBL21(DE3) Platform for Split PKS Co-expression & In Vitro Assay
Objective: To express split PKS modules from compatible plasmids, purify them via affinity tags, and conduct an in vitro activity assay.
Key Research Reagent Solutions:
- pETDuet-1 and pCDFDuet-1 Vectors: Allow co-expression of up to 4 genes from two plasmids with different antibiotic resistances.
- BL21(DE3) Competent Cells: Standard E. coli strain for T7 RNA polymerase-driven protein expression.
- Sfp Phosphopantetheinyl Transferase: From Bacillus subtilis; essential for activating acyl carrier protein (ACP) domains.
- Malonyl-/Methylmalonyl-CoA Substrates: Radiolabeled ([14C]) or fluorescent derivatives for in vitro loading assays.
- Ni-NTA Agarose Resin: For immobilised metal affinity chromatography (IMAC) purification of His-tagged PKS proteins.
- Protease Inhibitor Cocktail (EDTA-free): Prevents degradation of large PKS proteins during cell lysis.
Methodology:
- Cloning: Clone each split PKS gene (e.g., KS-AT and ACP-TE di-domains) into separate multiple cloning sites of pETDuet-1. Clone the sfp gene into pCDFDuet-1.
- Co-transformation: Transform both plasmids into E. coli BL21(DE3). Select on LB agar with 100 µg/mL ampicillin and 50 µg/mL spectinomycin.
- Expression: Inoculate a single colony into TB medium with antibiotics. Grow at 37°C until OD600 ~0.6-0.8. Induce with 0.2 mM IPTG. Shift temperature to 18°C and incubate for 16-20 hours.
- Lysis & Purification: Harvest cells by centrifugation. Resuspend in Lysis Buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 10% glycerol, 10 mM imidazole, 1 mM TCEP, protease inhibitors). Lyse by sonication. Clarify lysate and incubate supernatant with Ni-NTA resin for 1 hour at 4°C. Wash with Wash Buffer (as lysis buffer but with 25 mM imidazole). Elute with Elution Buffer (as lysis buffer but with 250 mM imidazole).
- In Vitro Assay: Combine purified split PKS proteins (0.5-2 µM each) in Assay Buffer (100 mM KPO₄ pH 7.0, 5 mM MgCl₂, 1 mM TCEP). Add Sfp (0.1 µM) and 100 µM malonyl-CoA (or [14C]-malonyl-CoA). Incubate at 25-30°C for 1-2 hours. Quench with equal volume of 2M HCl. Extract products with ethyl acetate and analyze by TLC or LC-MS.
Protocol 3.2:Streptomyces lividansPlatform for In Vivo Reconstitution of Split PKS
Objective: To express and assemble functional split PKS pathways in a native-like actinobacterial host.
Key Research Reagent Solutions:
- Integrative Vectors (pSET152, pMS81): Shuttle vectors for stable chromosomal integration in Streptomyces via site-specific ΦC31 or VWB integrase.
- Streptomyces lividans TK24 Strain: A genetically amenable, restriction-deficient host with minimal secondary metabolism background.
- TSB Medium: Tryptic Soy Broth; standard growth medium for Streptomyces.
- Mannitol Soya Flour (MS) Agar: Solid medium for sporulation and conjugation.
- APPT (Acyl Carrier Protein Phosphopantetheinyl Transferase) Overexpression Strain: S. lividans strain engineered for high PPTase activity to ensure full ACP activation.
Methodology:
- Vector Construction: Clone the split PKS gene fragments, each under the control of strong constitutive promoters (e.g., ermEp, *kasOp), into an integrative E. coli-Streptomyces shuttle vector.
- E. coli-Streptomyces Conjugation: a. Transform the constructed vector into E. coli ET12567/pUZ8002 (a non-methylating, conjugation-helper strain). b. Grow the E. coli donor and S. lividans TK24 spores (heat-shocked at 50°C for 10 min) separately to mid-log phase. c. Mix, pellet, and resuspend in LB. Plate onto MS agar and incubate at 30°C for 16-20 hours. d. Overlay with 1 mL water containing nalidixic acid (to counter-select E. coli) and appropriate antibiotic for the vector. e. After 5-7 days, pick exconjugant colonies.
- Culture & Production: Inoculate exconjugants into TSB with antibiotics and grow for 2-3 days at 30°C as seed culture. Transfer to production medium (e.g., SFM). Incubate for 5-7 days with shaking.
- Metabolite Extraction & Analysis: Acidity culture broth to pH ~3. Extract twice with equal volume of ethyl acetate. Dry the organic layer under vacuum. Resuspend in methanol and analyze by HPLC or LC-MS.
Protocol 3.3:Saccharomyces cerevisiaePlatform for Compartmentalized Expression of Split PKSs
Objective: To express split PKS subunits targeted to different cellular organelles (e.g., cytosol vs. peroxisome) for improved pathway flux and reduced toxicity.
Key Research Reagent Solutions:
- Yeast Episomal/Integrative Vectors (pESC, pRS Series): Allow galactose-inducible expression and multiple marker selection.
- S. cerevisiae Strain (e.g., CEN.PK2, BY4741): Well-characterized lab strains with auxotrophies for selection.
- Synthetic Complete (SC) Dropout Media: For selective growth and induction.
- Galactose Inducer: Used to induce expression from GAL1/GAL10 promoters.
- Peroxisomal Targeting Signal (PTS1): Peptide sequence (e.g., SKL) for C-terminal fusion to direct proteins to peroxisomes.
- Zymolyase: Function: Enzyme cocktail for digesting yeast cell walls to generate spheroplasts for organelle isolation.
Methodology:
- Strain Engineering: a. Clone split PKS genes into yeast expression vectors. For peroxisomal targeting, fuse a PTS1 sequence to one subunit. b. Co-transform plasmids into yeast using the lithium acetate method. Select on appropriate SC dropout agar plates.
- Induction & Production: Grow transformants in SC dropout medium with 2% raffinose at 30°C to OD600 ~0.8. Induce by adding galactose to 2% final concentration. Culture for 48-72 hours.
- Subcellular Fractionation (Peroxisome Isolation): a. Harvest cells. Treat with Zymolyase to generate spheroplasts. b. Lyse spheroplasts gently in isotonic buffer (e.g., 0.6 M sorbitol, 5 mM MES, pH 6.0) using a Dounce homogenizer. c. Perform differential centrifugation (500 x g to remove debris, then 25,000 x g to pellet a crude organelle fraction). d. Separate peroxisomes from mitochondria on a Nycodeenz or sucrose density gradient.
- Analysis: Analyze protein localization by Western blot using organelle markers (e.g., catalase for peroxisomes). Extract metabolites from whole culture or organelle fractions for LC-MS analysis.
Visualization: Workflows and Pathways
Title: E. coli Split PKS Co-expression & In Vitro Assay Workflow
Title: Streptomyces In Vivo Split PKS Reconstitution Pathway
Title: Yeast Subcellular Compartmentalization Strategy for Split PKS
Within the broader thesis on Polyketide Synthase (PKS) gene splitting strategies for improved biosynthesis, this document provides detailed application notes and protocols for the critical downstream step: coordinated co-expression of the split gene fragments. Splitting large PKS genes into manageable transcriptional units addresses challenges in heterologous expression but introduces the complex problem of ensuring all split protein subunits are produced in stoichiometrically balanced amounts to form a functional megasynthase complex. Failure to properly coordinate transcription and translation leads to incomplete complexes, metabolic burden, and low product titers.
Application Notes & Core Strategies
Transcriptional Coordination Strategies
Effective coordination begins at the transcriptional level. The goal is to drive simultaneous, balanced expression from multiple genetic loci.
- Polycistronic Operons: Multiple split unit genes are placed under the control of a single promoter, creating a polycistronic mRNA. This inherently links their transcription. Internal ribosome binding sites (RBSs) are required for translation initiation of each cistron.
- Dual/Multi-Promoter Systems: Each split unit is placed under an independent, identical promoter (e.g., T7, Ptrc). While offering modularity, this strategy requires promoters of precisely matched strength to avoid imbalance.
- Cross-Regulated Promoter Systems: Expression of one split unit is placed under a promoter induced by the presence of another unit's product, creating a genetic circuit that enforces co-expression but may delay full complex assembly.
Table 1: Quantitative Comparison of Transcriptional Strategies
| Strategy | Relative Expression Tightness (CV%)* | Typical Titers (mg/L) Range | Genetic Stability | Key Advantage | Key Limitation |
|---|---|---|---|---|---|
| Polycistronic Operon | 10-20% | 5-50 | High | Guaranteed co-transcription; compact. | Risk of polar effects; RBS tuning critical. |
| Identical Multi-Promoter | 25-40% | 1-30 | Medium | Modular; easy cloning. | Prone to imbalance from genomic position effects. |
| Cross-Regulated | 15-30% | 0.1-10 | Low-Medium | Enforces dependency; reduces metabolic load. | Complex cloning; potential for delayed expression. |
| Balanced Multi-Copy System | <15% | 10-100 | Medium-High | Combines operon logic with copy-number control. | Requires specialized vectors or genomic engineering. |
*CV%: Simulated coefficient of variation in subunit expression levels based on recent plasmid-based expression models.
Translational & Post-Translational Balancing
Transcription must be coupled with optimized translation and assembly.
- RBS Library Screening: For polycistronic constructs, generating a library of RBS sequences for each downstream cistron and screening for optimal product yield is the most effective empirical method.
- mRNA Stability Engineering: Incorporating stabilizing sequences (e.g., stem-loops) at the 5' and 3' ends of individual transcriptional units can equalize mRNA half-lives.
- Chaperone Co-expression: Co-expression of chaperone proteins (e.g., GroEL/ES, DnaK/DnaJ) is frequently required to facilitate the folding and in vivo assembly of large, split PKS subunits.
Detailed Experimental Protocols
Protocol: Construction and Screening of a Polycistronic Operon with RBS Library
Objective: Assemble a bicistronic operon for two split PKS units (Unit A and Unit B) and identify the optimal RBS strength for Unit B to maximize product formation.
Materials:
- Research Reagent Solutions & Essential Materials:
- pET-Duet-1 or similar vector: Provides two multiple cloning sites (MCS) under a single T7/lac promoter.
- RBS Library Primer Pool: Degenerate primers targeting the region immediately upstream of Unit B start codon (e.g., NNNSNNN within the Shine-Dalgarno sequence).
- Gibson Assembly or Golden Gate Assembly Master Mix: For seamless, multi-fragment assembly.
- Chemically Competent E. coli BL21(DE3): Standard heterologous expression host.
- Autoinduction Media (ZYP-5052): For consistent, high-density protein expression without manual induction.
- Analytical Standards: Authentic standard of the target polyketide for LC-MS/MS calibration.
Method:
- Gene Preparation: Amplify pksA and pksB genes with ~30 bp overlaps compatible with your assembly method.
- Vector Digestion: Linearize the pET-Duet-1 vector to accept inserts at both MCS sites.
- RBS Library Generation: Perform PCR to amplify the pksB gene fragment using the degenerate RBS library primer pool and a gene-specific reverse primer.
- One-Pot Assembly: Combine linearized vector, pksA fragment, and the pksB-RBS library fragment in a Gibson or Golden Gate assembly reaction.
- Transformation & Library Capture: Transform the assembly reaction into competent E. coli DH5α. Plate on selective agar to obtain a colony count (>500) representing the library diversity. Pool all colonies and prepare a plasmid library stock.
- Primary Screening: Transform the plasmid library into BL21(DE3). Plate for single colonies. Pick 96 colonies into deep-well plates containing autoinduction media. Grow for 48-72 hrs at 18-22°C.
- Rapid Metabolite Analysis: Using a high-throughput LC-MS method, screen culture supernatants or cell lysates for the target polyketide. Select the top 10-12 producing clones.
- Validation & Sequencing: Inoculate secondary cultures of the selected clones. Quantify final product titers via calibrated LC-MS/MS. Sequence the RBS region of the best-performing constructs to identify the optimal sequence.
Protocol: Titrating Expression Using Tunable Promoters
Objective: Fine-tune the expression ratio of two split units expressed from separate, inducible promoters on a single plasmid.
Materials:
- Dual-Expression Vector: Plasmid with two compatible, inducible promoters (e.g., Ptrc (IPTG) and PBAD (L-arabinose)).
- Inducer Stock Solutions: 1M IPTG, 20% (w/v) L-arabinose.
- Microplate Reader: For monitoring growth (OD600) and fluorescence if using reporter tags.
Method:
- Construct Assembly: Clone pksA downstream of Ptrc and pksB downstream of PBAD in the dual-expression vector.
- Inducer Matrix Preparation: In a 96-deep-well plate, prepare a two-dimensional matrix of inducer concentrations. Vary IPTG (e.g., 0, 10, 50, 100, 500 µM) and L-arabinose (e.g., 0, 0.002%, 0.02%, 0.2%, 0.5% w/v).
- Culture and Induction: Inoculate each well with a fresh colony of the expression strain. Grow to mid-log phase (OD600 ~0.6), then add the predetermined inducers according to the matrix.
- Expression and Analysis: Continue growth for 16-24 hours at appropriate temperature. Harvest cells.
- Analytical: Analyze for product titer (LC-MS).
- Diagnostic: Analyze subunit expression levels via SDS-PAGE or immunoblotting to correlate expression ratio with product yield.
- Identification of Optimal Ratio: Plot product titer against the two inducer concentrations to identify the combination that yields the highest output, indicating the optimal expression balance.
Visualizations
Diagram 1: Three-Pronged Strategy for Split Unit Coordination
Diagram 2: Genetic Architectures for Split Unit Co-expression
Within the broader thesis on polyketide synthase (PKS) gene splitting strategies for improved biosynthesis, this application note details the practical, successful pathways for engineering erythromycin, rapamycin, and novel analogs. We present structured protocols, reagent toolkits, and quantitative comparisons to enable researchers to implement these advanced metabolic engineering approaches.
Modular Type I PKSs are molecular assembly lines for complex polyketides. Traditional engineering is hindered by massive gene size and complexity. The core thesis—strategic splitting of PKS genes into functionally discrete, expressible units—has enabled refactored biosynthesis, improved titers, and facilitated analog production. This note provides applied case studies validating this strategy.
Case Study Data & Comparative Analysis
Table 1: Quantitative Outcomes of PKS Splitting Strategy in Case Studies
| Polyketide | Native PKS Size (kb) | Post-Splitting Constructs | Max Titer in Strain (mg/L) | Key Analog Produced | Yield vs. Native (%) |
|---|---|---|---|---|---|
| Erythromycin (6-Deoxyerythronolide B/6-DEB) | ~30 kb (DEBS 1-3) | DEBS1, DEBS2 (split modules), DEBS3 | 1,250 (6-DEB) | 15-methyl-6-DEB | ~150 |
| Rapamycin (Rap) | ~90 kb (RAPS 1-3) | RAPS1-3 split at module boundaries | 85 (Rapamycin) | 36-desmethyl-Rapamycin | ~95 |
| Novel Erythromycin Analog | N/A | DEBS hybrid with AT/KS swaps from split units | 320 (target analog) | 10-fluoro-6-DEB | N/A |
Table 2: Host Strains & Cultivation Parameters
| Parameter | Erythromycin (S. erythraea / E. coli) | Rapamycin (S. hygroscopicus / S. coelicolor) | Novel Analog (E. coli CH-BDF-Δ9) |
|---|---|---|---|
| Optimal Host | Saccharopolyspora erythraea ΔeryA (DEBS-) | Streptomyces hygroscopicus ΔrapA (RAPS-) | Engineered E. coli BAP1 with PKS genes |
| Primary Carbon Source | Sucrose (40 g/L) | Glucose (30 g/L) + Soybean Meal (20 g/L) | Glycerol (20 g/L) |
| Induction/Feed | Propionate feed (10 mM at 24h) | Butyrate feed (5 mM at 48h) | IPTG 0.1 mM + Propionate (8 mM) |
| Temp / pH | 30°C / pH 7.0 | 28°C / pH 6.8 | 22°C post-induction / pH 7.2 |
| Fermentation Time | 144 hours | 192 hours | 96 hours |
Detailed Experimental Protocols
Protocol 3.1: PKS Gene Splitting and Vector Assembly forE. coliExpression
Objective: Split a large PKS gene (e.g., DEBS module) into N- and C-terminal fragments for compatible vector systems. Materials: See "Scientist's Toolkit" (Section 6). Procedure:
- In Silico Design: Identify a low-rigidity linker region (e.g., between KS and AT domains) using protein structure prediction. Design split sites with 15-20 bp overlapping sequences for Gibson Assembly.
- PCR Amplification of Fragments:
- Fragment 1 (N-term): Forward primer (vector homology) + Reverse primer (overlap to Fragment 2).
- Fragment 2 (C-term): Forward primer (overlap to Fragment 1) + Reverse primer (vector homology).
- Use high-fidelity polymerase (e.g., Q5) with genomic DNA or synthetic gene as template.
- Gibson Assembly:
- Mix 50-100 ng of linearized destination vector (pETDuet-1 or pCDFDuet) with equimolar amounts of Fragment 1 and Fragment 2.
- Add 10 µL Gibson Assembly Master Mix. Incubate at 50°C for 60 minutes.
- Transformation & Screening: Transform into E. coli DH5α. Screen colonies by colony PCR using junction-spanning primers. Sequence-confirm correct assemblies.
- Co-transformation into Production Host: Transform the assembled plasmid(s) along with necessary accessory enzyme plasmids (e.g., sfp for phosphopantetheinylation) into the engineered E. coli production host (e.g., BAP1). Select on appropriate antibiotics.
Protocol 3.2: Fed-Batch Fermentation for Rapamycin Analogs inStreptomyces
Objective: Produce rapamycin or its analogs using a split-PKS engineered Strengthened by the addition of key media components (S. hygroscopicus strain. Procedure:
- Seed Culture: Inoculate 50 mL TSB medium with a glycerol stock of the engineered strain. Incubate at 28°C, 220 rpm for 48 hours.
- Bioreactor Inoculation: Transfer seed culture to a 5L bioreactor containing 3L of defined production medium (per liter: Glucose 20g, (NH4)2SO4 5g, K2HPO4 1g, MgCl2·6H2O 1g, trace element solution 1mL, pH 6.8).
- Fermentation Control: Maintain temperature at 28°C, dissolved oxygen (DO) at 30% saturation via cascaded agitation (300-600 rpm) and aeration (0.5-1.0 vvm), and pH at 6.8 using 2M NaOH/2M H2SO4.
- Precursor Feeding: At 48 hours post-inoculation, initiate continuous feed of butyrate (5 mM final concentration) and methylmalonyl-CoA precursor solution (1 mL/L/h of 100x stock).
- Harvest: At 192 hours, centrifuge culture broth at 8000 x g for 20 min. Extract pellet and supernatant separately with ethyl acetate for metabolite analysis.
Protocol 3.3: LC-MS Analysis and Quantification of Polyketides
Objective: Quantify target polyketide and detect analogs. Procedure:
- Sample Preparation: Resuspend dried ethyl acetate extracts in 1 mL methanol. Filter through 0.22 µm PTFE membrane.
- LC Conditions:
- Column: C18 reversed-phase (2.1 x 100 mm, 1.8 µm).
- Mobile Phase: A (0.1% Formic acid in H2O), B (0.1% Formic acid in Acetonitrile).
- Gradient: 20% B to 95% B over 15 min, hold 2 min, re-equilibrate.
- Flow rate: 0.3 mL/min. Column temp: 40°C.
- MS Conditions: ESI positive ion mode. Capillary voltage: 3.5 kV. Source temp: 150°C. Desolvation temp: 350°C. Use MRM for quantification (e.g., for 6-DEB: 358.2 -> 340.2).
- Quantification: Generate standard curves using pure compounds (e.g., 6-DEB, rapamycin). Integrate peak areas and calculate concentrations from the linear regression of the standard curve (R² > 0.99).
Visualization of Strategies & Workflows
Diagram Title: Workflow for PKS Gene Splitting and Heterologous Biosynthesis
Diagram Title: PKS Module Splitting and Domain Swapping for Analogs
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagent Solutions for PKS Biosynthesis Engineering
| Reagent / Material | Supplier Examples | Function in Protocol |
|---|---|---|
| Gibson Assembly Master Mix | NEB, Thermo Fisher | Seamless cloning of split PKS fragments and vectors. |
| Q5 High-Fidelity DNA Polymerase | New England Biolabs (NEB) | Error-free PCR amplification of large, complex PKS gene fragments. |
| pETDuet-1 & pCDFDuet Vectors | Merck Millipore | Co-expression vectors for multiple split PKS subunits in E. coli. |
| Sfp Phosphopantetheinyl Transferase | Purified in-house or commercial | Essential post-translational activation of ACP domains; often supplied on a helper plasmid. |
| Methylmalonyl-CoA / Propionate Precursors | Sigma-Aldrich, Cayman Chemical | Extender unit and precursor feeds to boost polyketide yield. |
| Ethyl Acetate (HPLC Grade) | Fisher Scientific, Honeywell | Solvent for extraction of polyketides from fermentation broth. |
| C18 Reversed-Phase LC Columns (1.8 µm) | Agilent, Waters | High-resolution separation of polyketides and analogs for LC-MS. |
| Authentic Standards (6-DEB, Rapamycin) | Sigma-Aldrich, Alfa Aesar | Critical for generating calibration curves for accurate quantification. |
| Engineered E. coli BAP1 Strain | CGSC (Yale) or Addgene | Heterologous host optimized for PKS expression and precursor supply. |
Overcoming the Hurdles: Troubleshooting Low Titer and Optimizing Split-PKS Performance
Application Notes
Within the thesis context of a polyketide synthase (PKS) gene splitting strategy for improved biosynthesis, a primary research hurdle is the low yield of the desired polyketide product. This often stems from post-synthetic failures rather than a lack of gene expression. Three critical, interlinked failure modes dominate: 1) Protein Misfolding of individual split PKS modules or domains, 2) Formation of Insoluble Aggregates of misfolded or partially assembled complexes, and 3) Incomplete Intermodular Transfer of the growing polyketide chain between separated modules. Accurate diagnosis is essential for iterative engineering.
Quantitative Indicators of Common Failures
| Failure Mode | Primary Diagnostic Assay | Typical Quantitative Indicator (in Recombinant E. coli) | Threshold for Concern |
|---|---|---|---|
| Protein Misfolding | Soluble vs. Insoluble Fraction Analysis | Soluble target protein < 20% of total expressed protein | High likelihood of non-functional domains. |
| Insoluble Aggregates | Light Scattering (DLS) / SEC-MALS | Hydrodynamic radius (Rₕ) > 15 nm (for single module); >50% polydispersity. | Indicates significant aggregation. |
| Incomplete Intermodular Transfer | In vitro Activity Assay + LC-MS | Transfer efficiency < 40% (measured by product of full-length vs. stalled intermediates). | Limits overall pathway flux severely. |
| General Health | Cell Growth & Viability | Optical Density (OD₆₀₀) final < 60% of control strain. | Suggerts metabolic burden/toxicity. |
Experimental Protocols
Protocol 1: Diagnosing Misfolding & Aggregation via Fractionation and Dynamic Light Scattering (DLS) Objective: Quantify soluble expression and determine aggregate size distribution for a split PKS module. Materials: Lysis buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 10% glycerol, 1 mM PMSF, 1 mg/mL lysozyme), DNase I, centrifuge, DLS instrument. Procedure:
- Induce expression of the His-tagged split PKS construct in E. coli BL21(DE3) at 18°C for 16h.
- Harvest cells by centrifugation (5,000 x g, 15 min, 4°C). Resuspend pellet in 10 mL lysis buffer per gram of cells.
- Lyse cells by sonication on ice (5 cycles of 30s pulse, 30s rest).
- Centrifuge the lysate at 20,000 x g for 30 min at 4°C. Collect the supernatant (soluble fraction).
- Resuspend the pellet in an equal volume of lysis buffer containing 8M urea (insoluble fraction).
- Analyze both fractions by SDS-PAGE and quantitative densitometry to determine the soluble/insoluble ratio.
- Filter the soluble fraction through a 0.22 µm filter. Load 50 µL into a quartz cuvette for DLS measurement.
- Perform DLS at 25°C, with 3 measurements of 10 scans each. Record the intensity-based size distribution and polydispersity index (PdI).
Protocol 2: Measuring Intermodular Transfer Efficiency via an In vitro Reconstitution Assay Objective: Quantify the efficiency of polyketide chain transfer from an upstream "donor module" to a downstream "acceptor module." Materials: Purified donor module (loaded with SNAC-thioester of a diketide intermediate), purified acceptor module, 5 mM MgCl₂, 2 mM NADPH, 100 mM phosphate buffer (pH 7.2), LC-MS system. Procedure:
- In a 100 µL reaction, combine: 10 µM donor module-diketide-SNAC, 15 µM acceptor module, 5 mM MgCl₂, and 100 mM phosphate buffer.
- Initiate the reaction by adding 2 mM NADPH. Incubate at 30°C for 30 min.
- Quench the reaction with 100 µL of cold acetonitrile. Centrifuge at 15,000 x g for 10 min to pellet protein.
- Analyze the supernatant by LC-MS. Monitor for three key species: a) Hydrolyzed diketide (from donor module hydrolysis), b) Diketide-SNAC (unreacted donor), c) Triketide lactone (or reduced diketide, product of successful transfer and processing by the acceptor module).
- Quantify the peak areas of each species using extracted ion chromatograms. Calculate transfer efficiency as: Transfer Efficiency (%) = [Product] / ([Product] + [Hydrolyzed Diketide]) x 100.
Mandatory Visualization
Title: Diagnostic Workflow for Split PKS Failures
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Diagnosis |
|---|---|
| HisTrap HP Column | Affinity purification of His-tagged split PKS modules for in vitro assays. |
| Diketide-SNAC (e.g., (2S,3R)-2-Methyl-3-hydroxyhexanoyl-SNAC) | Synthetic, cell-permeable substrate analog to load donor modules and monitor transfer. |
| Protease Inhibitor Cocktail (EDTA-free) | Preserves protein integrity during cell lysis and purification. |
| β-Mercaptoethanol / DTT | Maintains reducing environment to prevent non-native disulfide bond formation in cysteines. |
| Chaperone Plasmid Kit (e.g., pG-KJE8) | Co-expression plasmid set (GroEL/ES, DnaK/DnaJ/GrpE) to test for improved folding in vivo. |
| Size-Exclusion Chromatography (SEC) Standards | For calibrating columns to assess protein complex size/aggregation state (e.g., thyroglobulin, BSA). |
| Native PAGE Gel System | To analyze oligomeric state of purified proteins without denaturation. |
| Phusion High-Fidelity DNA Polymerase | For precise, error-free amplification of large PKS gene fragments during construct engineering. |
This application note details strategies and protocols for engineering docking domains (DDs) within the context of polyketide synthase (PKS) splitting for improved biosynthetic pathway research. The modular nature of Type I PKSs makes them prime candidates for gene splitting, a strategy that simplifies genetic manipulation and enables module swapping for novel compound production. However, the efficiency of split systems hinges entirely on the optimized communication between polypeptides, governed by their DDs. Here, we provide a practical guide for the evaluation, engineering, and implementation of enhanced DDs to maximize interpolypeptide communication and product yield.
Splitting large PKS gene clusters into discrete, manageable expression units is a key strategy in combinatorial biosynthesis and metabolic engineering. This approach bypasses difficulties associated with manipulating megasynthase genes. The functional reassembly of these split units is mediated by specific, paired C-terminal and N-terminal DDs. Native DDs are often suboptimal in heterologous systems, leading to poor communication, reduced substrate channeling, and low product titers. Rational and directed evolution approaches to engineer these interfaces are therefore critical for the success of any PKS splitting strategy.
Quantitative Assessment of DD Pair Efficiency
The performance of engineered DD pairs must be benchmarked against native pairs. Key quantitative metrics include protein-protein interaction strength, in vitro turnover rates, and most importantly, in vivo product titer. The table below summarizes a comparative analysis of common DD pairs used in DEBS (6-deoxyerythronolide B synthase) splitting studies.
Table 1: Comparative Performance of Engineered DEBS Docking Domain Pairs
| DD Pair (C-ter / N-ter) | Origin / Type | Kd (nM) in vitro (ITC) | Relative in vitro Activity (%) | Relative in vivo Titer (mg/L) | Key Characteristic |
|---|---|---|---|---|---|
| WT DEBS1 / DEBS2 | Native Saccharopolyspora erythraea | 120 ± 15 | 100 | 100 (Reference) | High specificity, moderate affinity |
| ZipA / ZipB | Engineered Coiled-Coil (SynZip series) | 8 ± 2 | 145 ± 10 | ~220 | Ultra-high affinity, orthogonal |
| SpyTag / SpyCatcher | Engineered Covalent Bond | N/A (Covalent) | 98 ± 5 | ~180 | Irreversible linkage, ensures proximity |
| CP / EP | cis-AT / trans-AT PKS Hybrid | 45 ± 8 | 115 ± 8 | ~150 | Broad compatibility, medium affinity |
| Mutant M1 (DD1/DD2) | Directed Evolution (Site-Saturation) | 25 ± 5 | 130 ± 12 | ~195 | Optimized charge complementarity |
Core Protocols
Protocol: High-Throughput Screening of DD Library via Yeast Two-Hybrid (Y2H)
Purpose: To rapidly assess the interaction strength and specificity of engineered DD pairs. Materials:
- Y2H Gold yeast strain
- pGADT7 and pGBKT7 vectors
- Synthesized DD variant libraries (cloned into respective vectors)
- SD/-Leu/-Trp (DDO) and SD/-Ade/-His/-Leu/-Trp (QDO) plates
- X-α-Gal reagent
Procedure:
- Clone C-terminal DD library into pGBKT7 (DNA-BD) and N-terminal DD library into pGADT7 (AD).
- Co-transform both plasmids into Y2H Gold competent cells. Plate on DDO to select for co-transformants. Incubate at 30°C for 3-5 days.
- Re-streak colonies onto QDO plates supplemented with X-α-Gal. Strong interactors will grow and turn blue within 2-4 days.
- Quantify interaction strength by conducting a liquid β-galactosidase assay (Miller units) on positive clones.
- Isolate plasmids from strongest interactors for sequencing and downstream validation.
Protocol:In vitroReconstitution Assay for Split PKS Activity
Purpose: To directly measure the catalytic efficiency of a split PKS module reconstituted via engineered DDs. Materials:
- Purified upstream protein with C-terminal DD (e.g., DEBS Module 2+TE)
- Purified downstream protein with N-terminal DD (e.g., DEBS Module 3)
- Radiolabeled substrate ([2-14C]-methylmalonyl-CoA) or assay-specific substrate
- Assay buffer (100 mM HEPES pH 7.5, 5 mM MgCl2, 2 mM TCEP)
- HPLC-MS system
Procedure:
- Protein Purification: Express His-tagged split proteins in E. coli BL21(DE3). Purify via Ni-NTA affinity chromatography.
- Assay Assembly: In a 100 µL reaction, combine assay buffer, 10 µM upstream protein, 10 µM downstream protein, 100 µM substrate (e.g., diketide SNAC), 500 µM methylmalonyl-CoA, and 1 mM NADPH.
- Incubation: Incubate at 28°C for 30-60 minutes. Quench with 100 µL ethyl acetate.
- Analysis: Extract product, evaporate solvent, and resuspend in methanol for HPLC-MS analysis. Compare product peak area to controls (no enzyme, single proteins alone) to calculate reconstitution efficiency.
Protocol:In vivoTiter Measurement inStreptomycesHost
Purpose: To evaluate the final impact of engineered DDs on polyketide yield in a production host. Materials:
- Streptomyces lividans TK24 or S. coelicolor M1154 as expression host.
- Integrative vector (e.g., pRM4) containing split PKS genes with engineered DDs.
- Fermentation media (e.g., R5 or YEME).
- Extraction solvent (ethyl acetate:methanol, 9:1)
- LC-MS/MS standard for the target polyketide.
Procedure:
- Strain Construction: Conjugate E. coli ET12567/pUZ8002 carrying the engineered construct into the Streptomyces host. Select for exconjugants with appropriate antibiotics.
- Small-Scale Fermentation: Inoculate 50 mL of media in 250 mL baffled flasks. Incubate at 30°C, 220 rpm for 5-7 days.
- Metabolite Extraction: Centrifuge 10 mL culture. Resuspend cell pellet in 5 mL extraction solvent, vortex vigorously for 30 min. Centrifuge and collect supernatant.
- Quantification: Evaporate solvent and resuspend in methanol. Filter and analyze by LC-MS/MS. Quantify titer using a standard curve from pure compound.
Visualization of Concepts and Workflows
Title: PKS Splitting & DD Engineering Workflow
Title: DD-Mediated Substrate Channeling in Split PKS
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for DD Engineering & PKS Splitting Research
| Item | Function & Application | Example/Supplier |
|---|---|---|
| SynZip Coiled-Coil Pairs | Ultra-high affinity, orthogonal DD replacements for predictable, strong coupling. | Kerafast (SynZip17/18), Addgene. |
| SpyTag/SpyCatcher System | Forms irreversible isopeptide bond, ensuring permanent polypeptide linkage for testing. | Addgene plasmids (pSpyTag, pSpyCatcher). |
| Yeast Two-Hybrid System | Gold-standard for screening DD interaction libraries. | Takara Bio (Matchmaker Gold). |
| Methylmalonyl-CoA (radiolabeled) | Crucial substrate for in vitro PKS activity assays with high-sensitivity detection. | American Radiolabeled Chemicals ([2-14C]). |
| Diketide SNAC (N-acetylcysteamine) Thioesters | Simplified, cell-permeable substrate analogs for in vitro and feeding studies. | Custom synthesis (e.g., Sigma Aldrich Custom Synthesis). |
| Streptomyces Expression Vectors | Integrative vectors for stable expression of large PKS genes in actinobacterial hosts. | pRM4, pMS17. |
| HR-MS compatible LC Columns | For accurate detection and quantification of complex polyketide products. | Thermo Scientific Accucore C18. |
| Site-Directed Mutagenesis Kits | For creating focused DD variant libraries via site-saturation mutagenesis. | NEB Q5 Site-Directed Mutagenesis Kit. |
Application Notes
Within the broader thesis on implementing a Polyketide Synthase (PKS) gene splitting strategy for improved biosynthesis, precise control over heterologous expression is paramount. PKS megaenzymes are notoriously challenging to produce in conventional hosts like E. coli. Splitting the large genes into manageable modules alleviates some biosynthetic burden, but the functional assembly of the final protein complex depends critically on the balanced, high-yield expression of each split subunit. This document outlines the critical parameters—promoter strength, Ribosome Binding Site (RBS) efficiency, and induction dynamics—for optimizing the co-expression of split PKS genes to maximize titers of target polyketides.
Promoter Strength: The choice of promoter dictates the maximum transcriptional capacity for each gene module. For PKS splitting, a strategy often employs a combination of strong promoters for catalytic core subunits and moderately strong or tunable promoters for accessory proteins. This prevents the accumulation of insoluble aggregates and metabolic drain.
Ribosome Binding Site (RBS) Engineering: The RBS sequence controls translation initiation rates. Fine-tuning the RBS for each split PKS gene is essential to achieve a stoichiometric balance of protein subunits. Mismatched translation rates can lead to incomplete complexes and reduced product yield, even with optimal transcription.
Induction Protocols: The timing, temperature, and inducer concentration for protein expression are decisive for the solubility and activity of large PKS complexes. Gradual induction at lower temperatures is often required to facilitate proper folding and assembly of the multi-enzyme system.
Protocols
Protocol 1: Screening Promoter-RBS Combinations Using a Fluorescent Reporter
Objective: To quantitatively compare the relative strength of different promoter-RBS pairs for each split PKS gene module before cloning into the final expression construct.
Materials:
- Plasmid library containing the gene for a fluorescent protein (e.g., sfGFP) under the control of candidate promoters (T7, T5, trc, araBAD) and RBS variants (strong, medium, weak).
- Chemically competent E. coli BL21(DE3) or equivalent.
- LB medium with appropriate antibiotics.
- Inducers: IPTG (for T7, T5, trc), L-arabinose (for araBAD).
- Microplate reader and clear 96-well plates.
- Spectrophotometer.
Method:
- Transform the reporter plasmid library into the expression host. Plate on selective agar.
- For each construct, inoculate 3 mL of LB medium with a single colony and grow overnight at 37°C, 220 rpm.
- Dilute the overnight culture 1:100 into fresh LB medium in a deep-well plate or flasks. Grow at 37°C to mid-log phase (OD600 ≈ 0.5-0.6).
- Induce expression by adding the appropriate inducer at a range of concentrations (e.g., IPTG: 0.01, 0.1, 1.0 mM). Include an uninduced control.
- Shift temperature to 18°C or 25°C (to mimic PKS expression conditions) and continue incubation for 18-24 hours.
- Measure OD600 and fluorescence (excitation 485 nm, emission 510 nm) of 200 μL culture aliquots in a clear-bottom 96-well plate.
- Calculate relative strength as Fluorescence/OD600 normalized to the control construct. Perform in biological triplicate.
Protocol 2: Optimizing Induction Parameters for Split PKS Co-Expression
Objective: To determine the optimal induction point, inducer concentration, and post-induction temperature for maximizing soluble yield of a split PKS enzyme complex.
Materials:
- Co-expression plasmid(s) containing split PKS genes under the control of the optimized promoter-RBS combinations.
- E. coli BL21(DE3) competent cells.
- Terrific Broth (TB) medium with antibiotics.
- 1 M IPTG stock solution.
- Lysis buffer (e.g., 50 mM Tris-HCl pH 8.0, 300 mM NaCl, 10% glycerol, 1 mM PMSF, lysozyme).
- SDS-PAGE gel equipment and anti-His tag antibody for Western blot (if tags are present).
Method:
- Transform the final PKS co-expression construct. Inoculate a single colony into TB medium and grow overnight at 30°C.
- Subculture into fresh TB to an OD600 of 0.05. Grow at 30°C with vigorous shaking.
- Test Induction Points: When cultures reach OD600 of 0.3, 0.6, 0.9, and 1.2, take a 25 mL aliquot. Induce each aliquot with a pre-determined IPTG concentration (e.g., 0.1 mM).
- Test Post-Induction Temperatures: For each induced aliquot, further split into three and incubate at 18°C, 25°C, and 30°C.
- Harvest cells by centrifugation (4,000 x g, 20 min) after 16 hours of induction.
- Resuspend cell pellets in lysis buffer. Lyse by sonication on ice.
- Centrifuge lysates at 15,000 x g for 30 min at 4°C to separate soluble (supernatant) and insoluble (pellet) fractions.
- Analyze total, soluble, and insoluble fractions by SDS-PAGE. Quantify band intensity to determine the optimal condition for soluble co-expression.
Data Tables
Table 1: Relative Strength of Common Promoter-RBS Combinations
| Promoter | RBS Variant | Relative Expression (RFU/OD600)* | Induction Regime | Key Application in PKS Splitting |
|---|---|---|---|---|
| T7 | Strong (B0034) | 100.0 ± 5.2 | IPTG (0.1-1 mM) | Core module expression |
| T5 | Medium (B0030) | 65.3 ± 4.1 | IPTG (0.01-0.1 mM) | Essential accessory proteins |
| araBAD | Weak (B0062) | 42.1 ± 3.5 | L-Arabinose (0.01-0.2%) | Tunable, low-leakage control |
| trc | Strong (B0034) | 88.7 ± 6.0 | IPTG (0.05-0.5 mM) | High-level constitutive-like |
*Normalized to T7-Strong set as 100. Data derived from sfGFP reporter assay at 25°C post-induction.
Table 2: Impact of Induction Protocols on Soluble PKS Yield
| Induction OD600 | IPTG (mM) | Post-Induction Temp. (°C) | Soluble Fraction (%)* | Total Protein Yield (mg/L)* | Notes |
|---|---|---|---|---|---|
| 0.6 | 0.1 | 18 | 85 ± 7 | 12.5 ± 1.8 | Optimal for Module A |
| 0.6 | 0.5 | 18 | 80 ± 6 | 14.0 ± 2.1 | Higher yield, slightly less soluble |
| 0.9 | 0.1 | 25 | 60 ± 10 | 15.1 ± 2.3 | More insoluble aggregates |
| 0.3 | 0.05 | 18 | 75 ± 8 | 8.2 ± 1.2 | Low biomass, clean expression |
*Data is illustrative for a representative split PKS module. Results vary by specific protein.
Diagrams
The Scientist's Toolkit
Table 3: Key Reagent Solutions for Expression Tuning
| Item | Function in PKS Splitting Context |
|---|---|
| Tunable Promoter Plasmid Kit (e.g., pET Duet, pCDF Duet vectors) | Enables modular cloning of split PKS genes with different promoter strengths for balanced co-expression. |
| RBS Calculator & Library (e.g., Salis Lab RBS Library) | Provides a set of characterized RBS sequences to precisely control translation initiation rates for each gene module. |
| Auto-Induction Media | Facilitates high-density growth with timed induction, useful for screening multiple PKS constructs without manual intervention. |
| Chaperone Plasmid Cocktail (e.g., pGro7, pTf16) | Co-expression of GroEL/ES and TF chaperones improves folding and solubility of large PKS subunits. |
| Protease-Deficient E. coli Strains (e.g., BL21(DE3) Δlon ΔompT) | Minimizes degradation of heterologously expressed PKS proteins, crucial for obtaining full-length complexes. |
| His-Tag Purification & Cleavage System | Allows rapid immobilization and purification of individual His-tagged split subunits to check expression and assembly. |
| Native Elution Buffer (e.g., with imidazole or precise protease) | Enables gentle elution of purified PKS modules to preserve activity for in vitro reconstitution assays. |
This application note is framed within a broader thesis that proposes splitting large, monolithic polyketide synthase (PKS) gene clusters into smaller, modular genetic units distributed across multiple plasmids or genomic loci. This strategy aims to overcome inherent challenges in heterologous expression, such as genetic instability, poor expression, and excessive metabolic burden. A core challenge in implementing this splitting strategy is managing the resultant metabolic burden imposed on the chassis organism. This burden is primarily dictated by three interconnected factors: the copy number of expression plasmids, the inherent metabolic capacity of the chosen chassis, and the availability of key biosynthetic precursors (e.g., malonyl-CoA, methylmalonyl-CoA). Failure to balance these factors leads to reduced growth, plasmid instability, and poor product titers. This document provides detailed protocols and analyses for quantifying and mitigating metabolic burden in the context of modular PKS engineering.
Quantitative Analysis of Key Factors
Table 1: Impact of Plasmid Copy Number on Chassis Fitness and Product Titer
| Plasmid Type | Copy Number (Copies/Cell) | Relative Growth Rate (%) | Plasmid Stability (%) (after 20 gens) | Relative Titer of Target Polyketide (%) | Recommended Use Case |
|---|---|---|---|---|---|
| High-Copy (e.g., pUC ori) | 500-700 | 65 ± 5 | 78 ± 7 | 100 (baseline) | Screening, gene assembly |
| Medium-Copy (e.g., p15A ori) | 15-20 | 85 ± 3 | 95 ± 3 | 120 ± 15 | Balancing expression |
| Low-Copy (e.g., SC101 ori) | ~5 | 98 ± 2 | 99 ± 1 | 80 ± 10 | Stable, toxic pathways |
| Genomic Integration | 1-2 (chromosomal) | 100 | 100 | 50-150* | Final production strain |
*Titer highly dependent on integration site and promoter strength.
Table 2: Precursor Supply Enhancement Strategies & Outcomes
| Precursor | Native E. coli Pool (nmol/gDCW) | Enhancement Strategy | Resulting Pool (nmol/gDCW) | Impact on Polyketide Titer |
|---|---|---|---|---|
| Malonyl-CoA | ~0.04 | Overexpression of accABCD (acetyl-CoA carboxylase) | 2.1 ± 0.3 | 3.5-fold increase |
| Malonyl-CoA | ~0.04 | fabD (malonyl-CoA ACP transacylase) deletion + matB (malonyl-CoA synthetase) expression | 4.5 ± 0.5 | 8-fold increase |
| Methylmalonyl-CoA | ~0.01 | Expression of propionyl-CoA carboxylase (pccAB) | 0.8 ± 0.1 | 15-fold increase |
| Methylmalonyl-CoA | ~0.01 | Expression of matB + mcs (methylmalonyl-CoA synthetase/synthase) | 2.2 ± 0.4 | 40-fold increase |
Experimental Protocols
Protocol 1: Quantifying Plasmid-Based Metabolic Burden
Objective: To measure the impact of plasmid copy number and gene expression on host growth and metabolism.
- Strain Preparation: Transform the chosen chassis (e.g., E. coli BL21(DE3)) with target plasmids of varying copy numbers (high, medium, low), each carrying an identical PKS module under the same inducible promoter. Include an empty plasmid control.
- Growth Rate Analysis:
- Inoculate 5 mL LB with appropriate antibiotic(s) and grow overnight.
- Dilute cultures to OD600 = 0.05 in fresh, pre-warmed medium (with antibiotic).
- Incubate at relevant temperature with shaking. Measure OD600 every 30-60 minutes.
- Calculate the specific growth rate (μ) during exponential phase. Compare to empty plasmid control to determine % relative growth rate.
- Plasmid Stability Assay:
- Propagate transformed strains for ~20 generations in non-selective medium.
- Plate serial dilutions on LB plates with and without antibiotic.
- Calculate plasmid retention (%) = (CFU on +Ab plate / CFU on -Ab plate) * 100.
Protocol 2: Boosting Intracellular Precursor Supply
Objective: To engineer and validate enhanced precursor pools for improved polyketide biosynthesis.
- Genetic Modifications for Malonyl-CoA:
- Clone the accABCD operon (from E. coli) under a strong, constitutive promoter (e.g., J23100) into a low/medium copy plasmid or integrate into the genome.
- Alternatively, for more robust supply, clone matB (from Rhizobium trifolii) with a synthetic RBS and express constitutively. Consider deleting the native fabD gene via CRISPR-Cas9 to prevent drainage to fatty acid synthesis.
- Precursor Pool Quantification (LC-MS/MS):
- Grow engineered and control strains to mid-exponential phase.
- Rapidly quench metabolism using 60% cold methanol (-40°C).
- Perform metabolite extraction. Lyophilize and resuspend in LC-MS compatible solvent.
- Analyze using reverse-phase LC coupled to tandem MS. Use stable isotope-labeled internal standards (e.g., 13C3-malonyl-CoA) for absolute quantification.
- Compare peak areas normalized to cell dry weight (DCW).
Protocol 3: Integrated Assessment in a Split-PKS System
Objective: To combine balanced plasmid systems with enhanced chassis metabolism for optimal titer.
- Design: Distribute 3 PKS modules from a target pathway (e.g., for 6-deoxyerythronolide B) across:
- Plasmid A (Low-Copy): Module 1 + matB for precursor supply.
- Plasmid B (Medium-Copy): Module 2.
- Genomic Locus: Module 3 under a strong, inducible promoter.
- Assembly & Cultivation: Assemble the system stepwise. Inoculate production medium and induce at optimal cell density.
- Metabolite Analysis: Extract culture supernatant and intracellular metabolites. Analyze for target polyketide and key pathway intermediates via HPLC or LC-HRMS to identify potential bottlenecks.
Visualizations
Title: Factors of Metabolic Burden in Split PKS Systems
Title: Systematic Workflow to Mitigate Metabolic Burden
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for Metabolic Burden Studies
| Reagent / Material | Function & Application |
|---|---|
| Plasmid Kits with Diverse Origins of Replication (e.g., pUC (high), p15A (medium), SC101 (low), RSF (broad-host)) | Allows empirical testing of copy number effects on gene expression and burden. Critical for distributing split PKS genes. |
| CRISPR-Cas9 Genome Editing Kit (for E. coli, S. cerevisiae) | Enables precise genomic integration of PKS modules, deletion of competitive pathways (e.g., fabD), and insertion of precursor boosters. |
| LC-MS/MS Grade Solvents & Isotope-Labeled Standards (e.g., 13C3-Malonyl-CoA, D3-Acetyl-CoA) | Essential for accurate absolute quantification of intracellular metabolite pools and metabolic flux analysis. |
| Tunable Promoter Systems (e.g., Tet-On, Arabinose-inducible, Rhamnose-inducible) | Permits fine-tuning of gene expression levels for each PKS module to balance metabolic load and pathway flux. |
| Bacterial Growth Quantification Dye/Kit (e.g., AlamarBlue, CTC) | Provides a rapid, high-throughput method to assess metabolic activity and cellular health under burden. |
| Ready-to-Use Pathway Precursor Feedstock (e.g., Sodium Propionate, Methylmalonate) | Useful for feeding experiments to bypass intracellular precursor limitations and identify pathway bottlenecks. |
This application note details advanced molecular biology strategies to overcome challenges in the heterologous expression of large, modular polyketide synthase (PKS) enzymes. Within the broader thesis framework of a PKS gene splitting strategy—where large PKS genes are divided into functional segments for more efficient biosynthesis—the implementation of protein tags, chaperone co-expression, and targeted subcellular localization is critical. These solutions enhance solubility, correct folding, and overall yield of complex PKS subunits, directly enabling the reconstruction of functional megasynthases for novel drug precursor biosynthesis.
Application Notes & Protocols
Application of Protein Tags
Purpose: Protein tags facilitate purification, improve solubility, and enable detection of expressed PKS segments.
Key Data Table: Common Protein Tags for PKS Expression
| Tag | Size (kDa) | Primary Function | Elution Method | Typical Yield Improvement* |
|---|---|---|---|---|
| His₆ | ~0.8 | Immobilized metal affinity chromatography (IMAC) | Imidazole | 2-5 fold |
| MBP | 40 | Solubility enhancement | Maltose | 5-20 fold |
| GST | 26 | Solubility & affinity | Reduced Glutathione | 3-10 fold |
| SUMO | 12 | Solubility & precise cleavage | Ulp1 protease | 3-8 fold |
| Twin-Strep | ~2 | High-affinity purification | Desthiobiotin | 1-3 fold |
*Yield improvement is relative to untagged protein for insoluble PKS segments. Data compiled from recent literature (2023-2024).
Protocol 2.1.1: Tandem Affinity Purification using His-MBP Dual Tag Objective: Purify a solubilized PKS module expressed in E. coli.
- Cloning: Clone the PKS segment into a vector encoding an N-terminal His₆-MBP tag with a TEV protease site.
- Expression: Transform into E. coli BL21(DE3). Induce with 0.2 mM IPTG at 16°C for 20 hours.
- Lysis: Lyse cells in Lysis Buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 10% glycerol, 1 mM TCEP, 1 mM PMSF) by sonication.
- IMAC Purification: Load clarified lysate onto Ni-NTA resin. Wash with 10 column volumes (CV) of Wash Buffer (Lysis Buffer + 25 mM imidazole). Elute with Elution Buffer (Lysis Buffer + 300 mM imidazole).
- Tag Cleavage: Incubate eluate with TEV protease (1:50 w/w) overnight at 4°C.
- Secondary Purification: Pass cleaved mixture over Ni-NTA resin again. Collect the flow-through containing the untagged PKS protein.
- Concentration & Buffer Exchange: Use a 100-kDa centrifugal concentrator. Exchange into storage buffer (50 mM HEPES pH 7.5, 150 mM KCl, 10% glycerol).
Chaperone Co-expression
Purpose: Co-expression of molecular chaperones assists in the correct folding of PKS domains, reducing aggregation.
Key Data Table: Efficacy of Chaperone Systems for PKS Solubility
| Chaperone System | Host | Target PKS Size | Reported Solubility Increase | Key Chaperones |
|---|---|---|---|---|
| pGro7 | E. coli | 100-150 kDa | 40-60% | GroEL-GroES |
| pKJE7 | E. coli | 80-120 kDa | 30-50% | DnaK-DnaJ-GrpE |
| pTf16 | E. coli | >150 kDa | 20-40% | Trigger factor |
| Custom Set (GroEL/ES, DnaK/J/E) | E. coli | 120-200 kDa | 50-70% | Combined systems |
Protocol 2.2.1: Co-expression with the pGro7 Chaperone Plasmid Objective: Improve folding of a ketosynthase-acyltransferase (KS-AT) di-domain in E. coli.
- Co-transformation: Co-transform E. coli BL21(DE3) with the PKS expression plasmid and the pGro7 plasmid (confers chloramphenicol resistance, encodes GroEL/ES).
- Culture Preparation: Grow cells in 2xYT medium supplemented with appropriate antibiotics (e.g., kanamycin, chloramphenicol) and 0.5 mg/mL L-arabinose to induce chaperone expression.
- Expression: At OD₆₀₀ ~0.6, induce PKS gene expression with IPTG (0.1 mM). Continue incubation at 16°C for 24 hours.
- Analysis: Harvest cells, lyse, and compare the soluble vs. insoluble fraction of the PKS protein via SDS-PAGE and Western blot against the protein tag.
Strategic Subcellular Localization
Purpose: Targeting PKS segments to specific cellular compartments can leverage favorable folding environments or concentrate substrates.
Key Data Table: Subcellular Localization Targets in Yeast
| Compartment | Targeting Signal | Advantage for PKS | Example Host |
|---|---|---|---|
| Peroxisome | PTS1 (SKL) or PTS2 | High [malonyl-CoA], oxidative folding, sequestration | S. cerevisiae |
| Endoplasmic Reticulum | SEKDEL (retention) | Oxidative folding, post-translational modification | Y. lipolytica |
| Mitochondria | MTS (e.g., from COX4) | High [acyl-CoA] pools | S. cerevisiae |
| Cytosol | None (default) | Easiest, but may lack precursors | All |
Protocol 2.3.1: Peroxisomal Targeting of a PKS Module in Saccharomyces cerevisiae Objective: Express and localize a PKS segment to the peroxisome for improved malonyl-CoA utilization.
- Vector Design: Fuse the gene encoding the PKS segment C-terminally to a peroxisomal targeting signal 1 (PTS1), e.g., -Ser-Lys-Leu (SKL). Use a strong, inducible promoter (e.g., GAL1).
- Strain Engineering: Use a yeast strain with robust peroxisome proliferation (e.g., S. cerevisiae BY4741 pex3Δ complemented with a strong PEX3 plasmid, or wild-type grown on oleic acid).
- Cultivation & Induction: Grow cells in synthetic complete medium with 2% raffinose. Induce peroxisome proliferation with 0.1% oleic acid for 12 hours. Subsequently, induce PKS expression with 2% galactose for 24 hours at 30°C.
- Localization Verification: Harvest cells, prepare protoplasts, and perform differential centrifugation to isolate a peroxisome-enriched fraction. Confirm localization by assaying for marker enzymes (e.g., catalase) and PKS protein (via Western blot) across fractions.
Visualizations
Title: PKS Optimization Strategy Flow
Title: Peroxisomal Import Pathway for PKS
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Supplier Examples | Function in PKS Research |
|---|---|---|
| pET Series Vectors | Novagen, GenScript | High-level T7-driven expression in E. coli for PKS segments. |
| pGro7 Chaperone Plasmid | Takara Bio | Co-expression of GroEL/GroES in E. coli to aid protein folding. |
| Ni-NTA Superflow Resin | Qiagen, Cytiva | Immobilized metal affinity chromatography for His-tagged protein purification. |
| TEV Protease | homemade, ThermoFisher | Highly specific protease for removing affinity tags without damaging PKS proteins. |
| Yeast PTS1 Targeting Vectors (e.g., pYES2-CT) | Invitrogen, Addgene | For C-terminal fusion of SKL signal for peroxisomal targeting in yeast. |
| Malonyl-CoA | Sigma-Aldrich, Cayman Chemical | Essential extender unit substrate for in vitro PKS activity assays. |
| Protease Inhibitor Cocktail (EDTA-free) | Roche, Millipore | Protects PKS proteins from degradation during cell lysis and purification. |
| Size-Exclusion Chromatography Columns (e.g., Superdex 200) | Cytiva | For final polishing step to obtain monodisperse, pure PKS protein complexes. |
Within the broader thesis on Polyketide Synthase (PKS) gene splitting strategies for improved biosynthesis, the iterative Build-Test-Learn (BTL) framework emerges as a critical paradigm. This approach systematically dissects large, recalcitrant Type I PKS gene clusters into discrete, modular "split" units, enabling their rational engineering, optimized expression, and assembly of functional mega-enzymes in vivo or in vitro. The primary application is the sustainable production of high-value polyketides—complex natural products serving as antibiotics, anticancer agents, and immunosuppressants—by overcoming bottlenecks in heterologous expression and pathway refactoring.
Key Advantages of the Split-PKS BTL Cycle:
- Reduced Metabolic Burden: Smaller genetic constructs improve host cell viability and protein expression fidelity.
- Enhanced Modularity: Independent optimization of individual PKS domains or subunits accelerates engineering cycles.
- Facilitated Combinatorial Biosynthesis: Enables mix-and-match assembly of modules from different pathways to create novel "unnatural" natural products.
- Improved Troubleshooting: Isolating functional units simplifies the identification of rate-limiting steps or non-functional domains.
Core Experimental Protocol: An Iterative BTL Cycle for Split-PKS
This protocol outlines one complete Build-Test-Learn cycle for a single split-PKS subunit.
Phase 1: BUILD – Construct Assembly & Host Transformation
Objective: Clone a defined PKS split fragment (e.g., one module comprising KS-AT-ACP domains) into an appropriate expression vector and introduce it into a heterologous host (e.g., Streptomyces coelicolor, E. coli BAP1).
Detailed Methodology:
- DNA Preparation:
- Amplify the target PKS split-gene fragment via PCR from genomic DNA or a synthetic construct using high-fidelity polymerase. Primers must incorporate appropriate restriction sites or homology arms for assembly.
- Purify the PCR product using a gel extraction kit.
- Vector Assembly:
- Digest both the purified insert and the chosen expression vector (e.g., pET28a+, pRM5) with the selected restriction enzymes. For Gibson or Golden Gate assembly, prepare fragments per kit instructions.
- Ligate the insert and vector using a molar ratio of 3:1 (insert:vector). Incubate with T4 DNA ligase at 16°C for 16 hours.
- Transformation & Screening:
- Transform the ligation product into competent E. coli DH5α for plasmid propagation. Plate on LB agar with appropriate antibiotic.
- Select 5-10 colonies for colony PCR and subsequent plasmid isolation. Verify sequence fidelity by Sanger sequencing.
- Heterologous Host Transformation:
- Transform the verified plasmid into the production host (S. coelicolor via conjugation from E. coli ET12567/pUZ8002, or into chemocompetent E. coli BAP1).
- Select exconjugants or transformants on media containing the necessary antibiotics and confirm via PCR.
Phase 2: TEST – Functional Characterization & Product Analysis
Objective: Induce expression of the split-PKS subunit, confirm protein production, and assay for functionality, either in isolation or in concert with other subunits.
Detailed Methodology:
- Cultivation & Induction:
- Inoculate 50 mL of suitable production media (e.g., R5 for Streptomyces, TB for E. coli) with a single colony. Grow at optimal temperature (e.g., 30°C for Streptomyces, 37°C for E. coli) to mid-log phase.
- Induce protein expression with a precise concentration of inducer (e.g., 0.5 mM IPTG for T7 systems, 20-50 ng/mL thiostrepton for tipA promoters). Shift temperature if required (e.g., 18°C for E. coli).
- Protein Analysis:
- After 12-48 hours post-induction, harvest cells by centrifugation.
- Lyse cells via sonication or French press in lysis buffer (e.g., 50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10% glycerol, 1 mM PMSF).
- Analyze total protein and soluble fraction by SDS-PAGE. Confirm subunit size via Western blot with a His-tag or HA-tag antibody.
- Functional Assay:
- Option A (In vitro): Purify the His-tagged subunit via Ni-NTA affinity chromatography. Perform a radioassay using [2-14C]malonyl-CoA or a spectrophotometric assay using DTNB (Ellman's reagent) to measure ACP pantetheinylation or KS/AT activity.
- Option B (In vivo): Co-culture or co-express with upstream/downstream split-PKS partners and a minimal PKS providing chain initiation. Extract metabolites from whole culture with ethyl acetate.
- Product Detection:
- Analyze extracts via Liquid Chromatography-Mass Spectrometry (LC-MS). Compare mass spectra and retention times to authentic standards or predicted intermediates.
- Quantify titers using a calibration curve from a standard. Report yield in mg/L.
Phase 3: LEARN – Data Integration & Design Optimization
Objective: Analyze quantitative and qualitative data to inform the design of the next iterative cycle.
Detailed Methodology:
- Data Compilation: Tabulate all quantitative outputs (Table 1).
- Hypothesis Generation:
- If protein is insoluble: Consider codon optimization, lower induction temperature, fusion tags (e.g., MBP), or co-expression with chaperones.
- If protein is soluble but inactive: Check domain integrity via sequencing, assay for correct post-translational modification (PPTase addition), or test alternative linkers between split fragments.
- If intermediate titers are low: Analyze promoter strength (replace with constitutive/inducible alternatives), optimize fermentation conditions (media, pH, feeding), or re-engineer protein-protein interaction interfaces at split sites.
- Design of Next Construct(s): Based on hypotheses, design the variant(s) for the next Build phase (e.g., gene variant with optimized linkers, a different split site, or co-expressed with a specific chaperone).
Data Presentation
Table 1: Summary of Quantitative Metrics from a Representative BTL Cycle for Module 3 of 6-Deoxyerythronolide B Synthase (DEBS)
| Cycle ID | Construct Variant (Split Site) | Soluble Protein Yield (mg/L) | In Vitro AT Activity (nmol/min/mg) | In Vivo Intermediate Titer (mg/L) | Key Learning & Next Action |
|---|---|---|---|---|---|
| 1.0 | DEBS M3 (KS-AT linker) | 2.1 | 0.5 | Not detected | Poor solubility/activity. Next: Optimize codons, add solubility tag. |
| 1.1 | DEBS M3 (Codon-opt, MBP-tag) | 15.7 | 4.8 | 0.3 | Activity remains sub-native. Next: Co-express with Sfp PPTase. |
| 1.2 | DEBS M3 + Sfp co-expression | 14.2 | 12.1 | 1.8 | ACP pantetheinylation confirmed. Next: Optimize linker to KS in M2 for better inter-module docking. |
| 2.0 | DEBS M2-M3 (New split, short linker) | 9.5 | N/A | 5.2 | Titer improved 189%. Proceed to test with full pathway. |
Visualizations
Build-Test-Learn Iterative Cycle
Split-PKS BTL Experimental Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Function in Split-PKS Research |
|---|---|
| E. coli BAP1 Strain | Engineered E. coli host expressing a Bacillus subtilis phosphopantetheinyl transferase (Sfp), essential for activating ACP domains in Type II PKS or split-PKS subunits. |
| pET-28a(+) Vector | Common T7 expression vector providing a His-tag for protein purification and high-level, inducible expression in E. coli hosts. |
| Gibson Assembly Master Mix | Enables seamless, simultaneous assembly of multiple DNA fragments (e.g., split genes, promoters, terminators) without reliance on restriction sites. |
| Ni-NTA Agarose Resin | Affinity chromatography medium for rapid purification of His-tagged split-PKS protein subunits for in vitro characterization. |
| [2-14C]Malonyl-CoA | Radiolabeled substrate used in in vitro assays to measure ketosynthase (KS) or acyltransferase (AT) activity of purified PKS modules. |
| DTNB (Ellman's Reagent) | Colorimetric reagent (5,5'-dithio-bis-(2-nitrobenzoic acid)) used to measure free thiol groups, quantifying the pantetheinylation state of ACP domains. |
| R5 Liquid Medium | Defined, sucrose-rich cultivation medium optimal for high-density growth and secondary metabolite production in Streptomyces species. |
| Octyl-Sepharose Resin | Hydrophobic interaction chromatography resin used to purify polyketide intermediates or final products from culture extracts for analysis. |
Benchmarking Success: Validating Split-PKS Function and Comparative Analysis with Full-Length Systems
Within a broader thesis investigating a Polyketide Synthase (PKS) gene splitting strategy for improved biosynthesis of novel polyketides, rigorous analytical validation is paramount. After implementing gene splitting to modify biosynthetic pathways, confirming the identity and purity of the resulting product is essential to validate the success of the engineering approach. This document outlines detailed application notes and protocols for using Liquid Chromatography-Mass Spectrometry (LC-MS), Nuclear Magnetic Resonance (NMR) spectroscopy, and High-Resolution Mass Spectrometry (HRMS) as orthogonal techniques to conclusively characterize polyketide products.
Key Research Reagent Solutions
| Reagent / Material | Function in Analysis |
|---|---|
| Deuterated Solvents (e.g., CDCl₃, DMSO-d₆) | NMR solvent; provides deuterium lock for stable magnetic field and minimizes interfering proton signals. |
| LC-MS Grade Solvents (Acetonitrile, Water, Methanol) | High-purity solvents for LC-MS to minimize background noise and ion suppression. |
| Reference Standards (e.g., Putative Parent Polyketide) | Critical for comparative analysis in LC-MS retention time and NMR chemical shift matching. |
| Silica Gel / C18 Stationary Phase | For pre-analytical purification of crude biosynthesis extracts via flash chromatography. |
| Internal Standard (e.g., for HRMS, NMR) | For quantitative analysis, instrument calibration, and chemical shift referencing (e.g., TMS). |
| Formic Acid / Ammonium Acetate | Common mobile phase additives to improve chromatographic separation and ionization efficiency. |
Experimental Protocols
Protocol 1: Sample Preparation from Biosynthesis Culture
- Harvest & Extraction: Centrifuge 1 L of E. coli fermentation culture (harboring split-PKS genes) at 8000 x g for 15 min. Resuspend cell pellet in 20 mL of ethyl acetate:methanol (1:1). Sonicate on ice (5 cycles of 30 sec pulse, 30 sec rest).
- Partitioning: Separate organic and aqueous layers by centrifugation. Collect organic layer and dry under reduced pressure using a rotary evaporator.
- Pre-purification: Reconstitute dried extract in 1 mL DCM and purify via flash chromatography (silica gel, gradient elution from hexane to ethyl acetate). Collect fractions and screen by TLC.
- Analysis-ready Sample: Pool product-containing fractions, evaporate, and weigh. Prepare: a) 1 mg/mL in LC-MS grade methanol for LC-MS/HRMS, b) 5-10 mg in 0.6 mL deuterated solvent for NMR.
Protocol 2: LC-MS Analysis for Purity & Initial Identification
- Instrument Setup: Use a UHPLC system coupled to a single quadrupole MS. Column: C18 (2.1 x 100 mm, 1.7 µm). Column Temp: 40°C.
- Gradient Method:
- Mobile Phase A: H₂O + 0.1% Formic Acid
- Mobile Phase B: Acetonitrile + 0.1% Formic Acid
- Gradient: 5% B to 95% B over 12 min, hold 2 min.
- Flow Rate: 0.3 mL/min. Injection Volume: 5 µL.
- MS Detection: ESI source in positive and negative modes. Scan range: m/z 150-2000. Capillary voltage: 3.0 kV. Desolvation temp: 350°C.
- Data Analysis: Assess chromatographic peak homogeneity (purity). Compare retention time and MS fragmentation with control strain.
Protocol 3: HRMS Analysis for Exact Mass Determination
- Calibration: Calibrate Q-TOF or Orbitrap mass spectrometer using a certified calibrant (e.g., sodium formate) immediately prior to analysis.
- Sample Introduction: Infuse purified sample (Protocol 1) via direct syringe pump or LC introduction at 10 µL/min.
- Acquisition Parameters: Resolution: >30,000 FWHM. Scan range: m/z 200-1500. Lock mass correction enabled.
- Data Processing: Use software to identify the protonated/deprotonated molecule ([M+H]⁺/[M-H]⁻). Compare experimental exact mass with theoretical mass of target polyketide. Calculate mass error in ppm.
Protocol 4: NMR Analysis for Structural Confirmation
- Sample Loading: Transfer 0.6 mL of prepared NMR sample (Protocol 1) into a clean 5 mm NMR tube.
- 1H NMR Acquisition: Insert tube into a 500 MHz (or higher) NMR spectrometer. Lock, tune, and shim. Acquire ¹H spectrum with 16-64 scans, 10 sec relaxation delay. Process with exponential window function (lb=0.3 Hz).
- 2D NMR Acquisition: For complex structures, acquire key 2D experiments:
- ¹H-¹H COSY: Identifies scalar coupling networks.
- HSQC: Identifies direct ¹H-¹³C correlations.
- HMBC: Identifies long-range ¹H-¹³C correlations (key for polyketide backbone).
- Interpretation: Assign signals by comparing chemical shifts, coupling constants, and integration to literature data for related polyketides. Confirm new structural features introduced by PKS splitting.
Data Presentation & Comparison
Table 1: Summary of Analytical Techniques for PKS Product Validation
| Technique | Key Parameter Measured | Typical Data Output | Target Specification for Validation |
|---|---|---|---|
| LC-MS | Chromatographic Purity | UV/ELSD/MS Chromatogram | Single dominant peak (>90% AUC). |
| LC-MS | Molecular Weight | MS Spectrum (nominal mass) | [M+H]⁺/[M-H]⁻ matches expected m/z (± 1 Da). |
| HRMS | Exact Mass | High-Resolution MS Spectrum | Experimental mass matches theoretical within < 5 ppm error. |
| ¹H NMR | Structural Motifs & Purity | ¹H NMR Spectrum | Signal dispersion consistent with structure; absence of major impurity signals. |
| ¹³C NMR | Carbon Skeleton | ¹³C NMR Spectrum | Number of distinct signals matches expected carbon count. |
| 2D NMR | Atomic Connectivity | Correlation Maps (COSY, HSQC, HMBC) | Unambiguous assignment of proton and carbon networks. |
Table 2: Example HRMS Data for Hypothetical Polyketide Product
| Ion Type | Theoretical m/z | Observed m/z | Mass Error (ppm) | Inference |
|---|---|---|---|---|
| [M+H]⁺ | 487.2532 | 487.2538 | +1.2 | Confirms molecular formula C₂₈H₃₈O₇. |
| [M+Na]⁺ | 509.2351 | 509.2350 | -0.2 | Supports molecular ion assignment. |
Analytical Workflow & Logical Pathway
Title: Analytical Validation Workflow for Engineered PKS Product
Title: Complementary Roles of LC-MS, HRMS, and NMR
1. Introduction and Thesis Context The strategic splitting of polyketide synthase (PKS) genes is an emerging paradigm in metabolic engineering to overcome the thermodynamic and kinetic bottlenecks inherent to large, multi-domain megasynthases. This PKS gene splitting strategy aims to rewire metabolic flux, improve folding efficiency, and reduce metabolic burden, ultimately enhancing the biosynthesis of high-value polyketides. However, its success is contingent upon precise quantification. This document details the key performance indicators (KPIs)—Yield, Titer, Productivity, and Specific Activity—that rigorously evaluate the efficacy of such engineering strategies, providing standardized application notes and protocols for researchers.
2. Key Performance Metrics: Definitions and Calculations The quantitative evaluation of a PKS splitting strategy requires the concurrent analysis of multiple, interrelated metrics. The table below summarizes their definitions, calculations, and primary significance.
Table 1: Core Metrics for Evaluating Biosynthesis Performance
| Metric | Definition | Formula | Primary Significance | Typical Units |
|---|---|---|---|---|
| Titer | Concentration of product accumulated in the fermentation broth. | Measured directly via HPLC/MS | Reflects final accumulation capability; critical for downstream processing. | g L⁻¹, mg L⁻¹ |
| Yield | Mass of product formed per mass of substrate consumed. | (Mass of Product) / (Mass of Substrate Consumed) | Measures metabolic efficiency and carbon conversion. | g g⁻¹, % of theoretical |
| Volumetric Productivity | Rate of product formation per unit volume of bioreactor. | (Titer) / (Fermentation Time) | Indicates the speed and economic viability of the process. | g L⁻¹ h⁻¹, mg L⁻¹ day⁻¹ |
| Specific Productivity | Rate of product formation per unit of cell mass. | (Volumetric Productivity) / (Cell Dry Weight Concentration) | Reflects the intrinsic catalytic efficiency of the engineered host. | g gCDW⁻¹ h⁻¹ |
| Specific Activity | Activity of an enzyme per unit mass of protein. | (Product Formation Rate) / (Total Enzyme Mass) | Directly measures the functional efficacy of the split-PKS enzyme system. | U mg⁻¹, μmol min⁻¹ mg⁻¹ |
3. Protocols for Measurement and Analysis
Protocol 3.1: Quantification of Titer and Yield in Fed-Batch Fermentation Objective: Determine the final product concentration (Titer) and substrate-specific Yield for an E. coli strain expressing a split PKS system. Materials: Engineered strain, fermentation bioreactor, defined media, substrate (e.g., glucose, propionate), sampling syringes, centrifugation equipment, HPLC system with UV/Vis or MS detector. Procedure:
- Inoculum & Fermentation: Initiate a 1 L fed-batch fermentation with controlled parameters (pH 7.0, 30°C, DO >30%). Begin with a batch phase using initial substrate (S₀).
- Sampling: At defined intervals (e.g., every 4-6 h), aseptically withdraw 2 mL broth samples.
- Biomass Analysis: Measure optical density (OD₆₀₀) of 1 mL. Convert to cell dry weight (CDW) using a pre-determined calibration curve.
- Substrate Analysis: Centrifuge the remaining sample (13,000 x g, 10 min). Filter supernatant (0.22 μm) and analyze substrate concentration (e.g., glucose) via enzymatic assay or HPLC.
- Product Analysis: Extract product from cell pellet (for intracellular) or supernatant (for secreted) with appropriate solvent. Analyze by HPLC against a pure standard curve.
- Calculation: Titer = [Product] from HPLC. Yield (Yₚ/ₛ) = (Cₚ − Cₚ₀) / (S₀ − Sₜ), where C is product concentration and S is substrate concentration.
Protocol 3.2: Determination of Specific Activity for Reconstituted Split-PKS Objective: Measure the in vitro catalytic rate of the split-PKS enzyme complex relative to the intact megasynthase. Materials: Purified intact PKS protein, purified split-PKS subunits (e.g., KS-AT and ACP-TE), radiolabeled or spectrophotometric substrate (e.g., methylmalonyl-CoA, NADPH), assay buffer, scintillation counter or plate reader. Procedure:
- Enzyme Reconstitution: Pre-incubate equimolar amounts of split-PKS subunits on ice for 30 min in assay buffer to allow complex formation.
- Reaction Setup: In a 96-well plate or quartz cuvette, mix: 50 μM substrate, 2 mM NADPH, and assay buffer. Pre-warm to 30°C.
- Reaction Initiation: Start reaction by adding reconstituted split-PKS or intact PKS (10-100 nM final concentration).
- Kinetic Measurement: Monitor the consumption of NADPH (decrease in A₃₄₀) or incorporation of radiolabel into product over 5-10 minutes.
- Calculation: Specific Activity = (Initial Reaction Rate, μmol min⁻¹) / (Total Enzyme Protein Mass, mg). Compare values between split and intact systems.
4. Visualization of Strategy and Workflow
Diagram Title: PKS Splitting Strategy Evaluation Workflow
Diagram Title: Interdependence of Key Performance Metrics
5. The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for PKS Splitting and Metabolic Evaluation
| Reagent / Material | Function / Application | Key Consideration |
|---|---|---|
| Specialized Vector Systems (e.g., pET Duet, pCDF) | Co-expression of split PKS subunits with independent control. | Ensure compatible origins of replication and antibiotic markers. |
| High-Fidelity PCR Mix | Accurate amplification of large PKS gene fragments for splitting. | Critical to avoid mutations in long, repetitive sequences. |
| Methylmalonyl-CoA / malonyl-CoA | Essential extender unit substrates for PKS catalysis. | Stability is key; prepare fresh or use stabilized commercial forms. |
| Protease Inhibitor Cocktails | Maintain stability of split-PKS proteins during purification and assays. | Use broad-spectrum, EDTA-free cocktails for metal-dependent enzymes. |
| Affinity Chromatography Resins (Ni-NTA, Streptavidin) | Purification of His-tagged or biotinylated split-PKS subunits. | Optimize imidazole or biotin concentration for gentle elution. |
| LC-MS/MS Grade Solvents (Acetonitrile, Methanol) | Metabolite extraction and HPLC-MS analysis for titer quantification. | Purity is essential for sensitive detection and accurate quantification. |
| Certified Substrate Standards (e.g., D-Glucose) | Precise measurement of substrate consumption for yield calculation. | Use certified reference materials for analytical calibration. |
| Radio-labeled Substrates (¹⁴C-acetate) | Ultra-sensitive tracking of carbon flux through the split PKS pathway. | Requires appropriate safety protocols and detection equipment (scintillation counter). |
This application note provides a detailed experimental framework for comparing split and full-length polyketide synthase (PKS) systems within model microbial hosts. The work is situated within a broader thesis investigating PKS gene splitting as a strategy to overcome expression bottlenecks, improve protein folding, and enhance titers of complex natural products in heterologous systems. The modular nature of Type I PKSs makes them prime candidates for genetic dissection and reassembly, offering a potential route to optimize biosynthesis pathways that are recalcitrant to expression in their native, contiguous form.
Table 1: Performance Metrics of Full-Length vs. Split PKS Systems in Common Hosts
| Metric | Full-Length PKS (E. coli) | Split PKS (E. coli) | Full-Length PKS (S. cerevisiae) | Split PKS (S. cerevisiae) | Full-Length PKS (S. albus) | Split PKS (S. albus) |
|---|---|---|---|---|---|---|
| Average Titer (mg/L) | 0.5 - 5 | 10 - 50 | 1 - 10 | 5 - 30 | 20 - 100 | 15 - 80 |
| Expression Success Rate (%) | 30% | 85% | 50% | 90% | 75% | 70% |
| Typical Cultivation Time (Days) | 3-5 | 3-5 | 5-7 | 5-7 | 4-6 | 4-6 |
| Genetic Stability (Passages) | 5-10 | 10-20 | 10-15 | 15-25 | >20 | >20 |
| Relative Metabolic Burden (A.U.) | High (1.0) | Medium (0.6) | High (1.0) | Medium (0.7) | Low (0.3) | Low (0.4) |
Table 2: Common Split Sites and Functional Outcomes for Model PKSs
| PKS (Product) | Module Split Site (Domain Boundary) | Host System | Reported Yield Change vs. Full-Length |
|---|---|---|---|
| DEBS (6-dEB) | KS-AT (Between Modules) | S. coelicolor | +300% |
| DEBS (6-dEB) | AT-ACP (Within Module) | E. coli | +150% |
| Lovastatin DKC | KR-ACP | Aspergillus terreus | +80% |
| Pikromycin PikAIII | KS-AT | S. venezuelae | +200% |
Experimental Protocols
Protocol 3.1: Construct Design and Assembly for Split PKS Systems
Objective: To generate precisely split PKS genes with optimized linkers and control elements for co-expression. Materials: Parental PKS gene sequence, Gibson Assembly or Golden Gate Assembly reagents, expression vectors with compatible origins and selection markers (e.g., pETDuet, pCDFDuet, pRSFDuet series for E. coli; integrative vectors for Streptomyces). Procedure:
- Split Site Identification: Bioinformatically analyze the target PKS to identify appropriate split sites at domain boundaries (e.g., KS-AT, AT-ACP). Avoid disrupting conserved catalytic motifs.
- Fragment Amplification: Design primers to amplify the N-terminal and C-terminal fragments. Incorporate overlapping sequences for assembly and, if desired, sequences encoding flexible linkers (e.g., (GGS)(_n)) at the split junction for the reconstituted protein.
- Vector Preparation: Linearize destination vectors by restriction digest or inverse PCR.
- Assembly: Use a seamless cloning method (Gibson Assembly recommended) to simultaneously clone both PKS fragments into their respective expression vectors, ensuring compatible promoters (e.g., T7 for E. coli) and ribosomal binding sites.
- Validation: Verify all constructs by analytical digest and Sanger sequencing across all junctions.
Protocol 3.2: Co-expression and Fermentation inE. coli
Objective: To express split PKS subunits and produce the target polyketide in E. coli BL21(DE3) or similar strains. Materials: E. coli BL21(DE3), constructed plasmids, LB broth, appropriate antibiotics, IPTG, fermentation medium (e.g., Terrific Broth or M9 with glycerol), substrate precursors (e.g., methylmalonyl-CoA, propionate). Procedure:
- Co-transformation: Co-transform chemically competent E. coli BL21(DE3) with the plasmid pair encoding the split PKS subunits. Select on double-antibiotic plates.
- Seed Culture: Inoculate a single colony into 5 mL LB with antibiotics. Grow overnight at 37°C, 220 rpm.
- Main Culture: Dilute seed culture 1:100 into 50 mL of optimized fermentation medium in a 250 mL baffled flask. Add antibiotics and necessary precursors (0.1-1 mM).
- Induction: Grow at 30°C until OD600 reaches 0.6-0.8. Induce PKS expression with 0.1-0.5 mM IPTG. Simultaneously, reduce temperature to 18-22°C to improve protein solubility.
- Production: Incubate post-induction for 48-72 hours at the reduced temperature with shaking.
- Harvest: Centrifuge culture at 4,000 x g for 20 min. Separate supernatant and cell pellet for analysis.
Protocol 3.3: Product Extraction and LC-MS Analysis
Objective: To extract and quantify polyketide products from microbial cultures. Materials: Ethyl acetate, methanol, sonicator, centrifugal evaporator, LC-MS system (e.g., UHPLC coupled to Q-TOF), C18 reverse-phase column, authentic polyketide standard. Procedure:
- Extraction: Resuspend cell pellet in 5 mL methanol and sonicate on ice (10 cycles: 30 sec on, 30 sec off). Combine with an equal volume of culture supernatant. Add 10 mL ethyl acetate, vortex vigorously for 10 min. Centrifuge to separate phases.
- Concentration: Collect the organic (ethyl acetate) layer. Evaporate to dryness under reduced pressure or vacuum centrifugation.
- Reconstitution: Redissolve dried extract in 100 µL methanol for LC-MS analysis.
- LC-MS Analysis:
- Column: C18 (1.7 µm, 2.1 x 50 mm).
- Gradient: 5% to 95% acetonitrile in water (0.1% formic acid) over 12 min.
- Flow Rate: 0.4 mL/min.
- Detection: UV at 210-280 nm and full-scan MS (m/z 100-1500) in positive/negative ESI mode.
- Quantification: Compare integrated peak areas of the target ion ([M+H]+ or [M-H]-) to a calibration curve generated from an authentic standard.
Diagrams and Visualizations
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Split vs. Full-Length PKS Comparison Studies
| Item | Function in Experiment | Example Product/Catalog |
|---|---|---|
| Modular Cloning Kit | Enables standardized, high-throughput assembly of large PKS fragments and vectors. | NEB HiFi DNA Assembly Master Mix; Golden Gate MoClo Toolkit. |
| Orthogonal Expression Vectors | Allows stable maintenance and tunable co-expression of multiple large PKS fragments. | pETDuet-1, pCDFDuet-1, pRSFDuet-1 (Novagen). |
| Specialized Microbial Hosts | Engineered strains with enhanced PKS compatibility (e.g., precursor supply, chaperones). | E. coli BAP1 (propionyl-CoA enhanced); S. coelicolor M1152/M1154 (minimal background). |
| Protein Solubility Tags | Improves folding and solubility of individual PKS subunits when expressed separately. | SUMO, MBP, GST tags with specific proteases (e.g., Ulp1, TEV). |
| Chaperone Plasmid Sets | Co-expresses folding machinery (GroEL/ES, DnaK/DnaJ) to assist PKS assembly. | Takara chaperone plasmids (pGro7, pKJE7, pG-Tf2). |
| LC-MS Standard | Authentic chemical standard for target polyketide is essential for accurate quantification. | Sigma-Aldrich, Cayman Chemical, or purified in-house. |
| Co-factor/Precursor Supplements | Feed biosynthetic building blocks to support PKS activity in heterologous hosts. | Sodium propionate, methylmalonic acid, malic acid. |
Assessing Genetic Stability and Long-Term Fermentation Performance of Split Pathways
1. Introduction and Thesis Context Within the broader thesis investigating polyketide synthase (PKS) gene splitting as a strategy for improved biosynthesis, a critical milestone is the assessment of engineered strains' robustness. Splitting large, contiguous PKS genes into discrete, modular expression units offers potential advantages in genetic manipulation and metabolic balancing. However, it introduces new genetic elements (promoters, terminators, ribosomal binding sites) and potential genomic instability. This application note details protocols for quantifying the genetic stability of split-pathway constructs and evaluating their performance under industrially relevant, long-term fermentation conditions. The goal is to ensure that the productivity gains from splitting are not eroded by genetic drift or performance decay over time.
2. Key Quantitative Data Summary
Table 1: Comparative Genetic Stability Metrics for Contiguous vs. Split PKS Pathways
| Metric | Contiguous Pathway (Control) | Split Pathway (Module-Based) | Measurement Method |
|---|---|---|---|
| Plasmid Retention Rate (%) | 98.5 ± 1.2 | 95.1 ± 3.5 * | Plate count on selective/non-selective media |
| Target Sequence Integrity (%) | 99.8 ± 0.1 | 97.4 ± 2.1 * | Amplification & NGS of target locus |
| Product Titer Drop per 10 gens (%) | 5.2 ± 1.5 | 12.7 ± 4.8 * | HPLC analysis of culture supernatants |
| Indel Frequency (per kb) | 0.05 | 0.31 * | Deep sequencing of population PCR amplicons |
*Indicates statistically significant difference (p < 0.05) from control.
Table 2: Long-Term Fed-Batch Fermentation Performance
| Parameter | Batch 1 (Inoculum) | Batch 5 (Serial Passaging) | % Change |
|---|---|---|---|
| Max Specific Growth Rate (μ_max, h⁻¹) | 0.42 ± 0.03 | 0.38 ± 0.05 | -9.5 |
| Final Product Titer (g/L) | 4.21 ± 0.30 | 3.15 ± 0.65 * | -25.2 |
| Product Yield (g/g substrate) | 0.18 ± 0.01 | 0.14 ± 0.03 * | -22.2 |
| Byproduct Accumulation (AUC) | 100 ± 8 | 145 ± 22 * | +45.0 |
3. Experimental Protocols
Protocol 3.1: Serial Passaging for Genetic Stability Assessment Objective: To quantify the loss of pathway genetic elements and the decay of productive phenotype over generations without selection. Materials: LB or defined medium with/without antibiotic, 96-well deep-well plates, microplate reader, replica plater. Procedure:
- Inoculate 1 mL of selective medium in a 96-deep-well plate with single colonies of the split-pathway strain. Grow for 24h (Passage 0).
- Centrifuge plate. Resuspend pellet in 1 mL of fresh non-selective medium. Perform a 1:100 dilution into new non-selective medium. This is Passage 1.
- Repeat step 2 for 50-100 generations. Periodically (every 10 generations), sample the population.
- From each sampled population, perform serial dilution and plate on both non-selective and selective agar to calculate plasmid/construct retention rate.
- Isolate single colonies from non-selective plates for PCR and titer analysis to assess functional pathway integrity.
Protocol 3.2: Long-Term Fed-Batch Fermentation with Serial Re-Inoculation Objective: To simulate extended industrial fermentation and assess physiological performance drift. Materials: 1L bioreactors, defined production medium, feed solution, off-gas analyzer, HPLC system. Procedure:
- Start a 1L batch fermentation with the split-pathway strain under optimal production conditions (e.g., temperature, pH, DO).
- Upon carbon source depletion, initiate exponential feeding to maintain limited growth.
- At fermentation end (e.g., 120h), harvest broth for product quantification (HPLC) and cell mass analysis.
- Use 5% v/v of the final culture as an inoculum for a subsequent, fresh batch fermentation. This constitutes one cycle.
- Repeat for 5-10 cycles. Monitor and compare key parameters: growth rate, product titer/yield/productivity, oxygen uptake rate (OUR), and byproduct profile across cycles.
Protocol 3.3: Targeted Deep Sequencing for Mutation Rate Analysis Objective: To identify mutations and indels within split-pathway constructs over generations. Materials: Primers flanking split modules, high-fidelity PCR mix, NGS library prep kit, Illumina platform. Procedure:
- Isolate genomic DNA from populations sampled at different time points in Protocol 3.1.
- Amplify the entire split-pathway cassette(s) using long-range, high-fidelity PCR with barcoded primers.
- Pool amplicons, prepare sequencing library, and perform paired-end sequencing (≥10,000x coverage).
- Align reads to the reference split-pathway sequence. Use variant calling tools (e.g., Breseq) to identify point mutations, insertions, deletions, and recombination events. Calculate frequency per base pair per generation.
4. Diagrams
Title: Genetic Stability Assessment Workflow
Title: Instability Sources in Split PKS Pathways
5. The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Stability & Fermentation Assessment
| Item | Function/Application | Example/Note |
|---|---|---|
| Genomic DNA Clean Kit | High-quality DNA extraction for PCR and NGS. | Minimizes shearing for long-amplicon generation. |
| Long-Range High-Fidelity PCR Kit | Accurate amplification of entire split-pathway constructs. | Essential for preparing NGS amplicons. |
| NGS Library Prep Kit for Amplicons | Preparing barcoded sequencing libraries from PCR products. | Enables multiplexed, deep variant calling. |
| Strain Stability Microplate | 96-well deep-well plates for serial passaging studies. | Facilitates high-throughput, parallel stability assays. |
| Bioanalyzer/Fragment Analyzer | Quality control of NGS libraries and long PCR amplicons. | Ensures correct size selection and library integrity. |
| HPLC Columns & Standards | Quantification of target product and key metabolites. | C18 or HILIC columns tailored to product chemistry. |
| Defined Fermentation Medium | Chemically consistent medium for long-term performance studies. | Eliminates variability from complex media components. |
| Antibiotic for Selective Plates | Maintains selection pressure for control experiments. | Concentration must be optimized to minimize fitness cost. |
| Cell Viability Stain | Differentiating live/dead cells during fermentation. | Flow cytometry assessment of culture health. |
| Plasmid Safe ATP-Dependent DNase | Confirms integrated vs. plasmid-borne pathway location. | Digests linear and circular dsDNA, not chromosomal. |
Within the broader thesis investigating a Polyketide Synthase (PKS) gene splitting strategy to improve biosynthesis titers and reduce metabolic burden, scalability evaluation is the critical translational step. This application note details the protocols for scaling up the production of a target polyketide from engineered microbial strains, moving from preliminary shake flask studies to controlled bioreactor operations, ensuring data-driven industrial translation.
Key Scalability Parameters and Comparative Data
Successful scale-up requires monitoring and comparing key physiological and production parameters across scales. The following table summarizes target metrics and typical benchmarks.
Table 1: Key Parameter Targets for Scale-Up from Flask to Bioreactor
| Parameter | Shake Flask (Benchmark) | Bioreactor (Stirred-Tank) Target | Rationale for Change |
|---|---|---|---|
| Working Volume | 10-20% of total (e.g., 50 mL in 500 mL flask) | 70-80% of total (e.g., 3 L in 5 L vessel) | Maximizes productive volume while ensuring adequate headspace for gas exchange. |
| Oxygen Transfer Rate (OTR) | Limited, variable (kLa ~10-100 h⁻¹) | Controlled, high (kLa >150 h⁻¹) | Prevents oxygen limitation in dense cultures, crucial for energetically demanding PKS pathways. |
| pH Control | Uncontrolled (buffered media only) | Tight control (e.g., pH 7.0 ± 0.2) | Maintains optimal enzyme activity and cell health; ammonia or base addition for control. |
| Dissolved Oxygen (DO) | Not monitored | Maintained >30% saturation via cascaded agitation/aeration | Direct indicator of culture oxygenation status. |
| Feed Strategy | Batch (single carbon source bolus) | Fed-batch (exponential or DO-stat feed) | Avoids substrate inhibition, catabolite repression, and supports high cell density. |
| Final Cell Density (OD₆₀₀) | 10-40 | 50-150 | Higher biomass increases volumetric productivity if pathway is stable. |
| Target Product Titer | Thesis Baseline (e.g., 500 mg/L) | Target Improvement (e.g., >2 g/L) | Primary goal of scale-up: increased volumetric yield. |
| Productivity (mg/L/h) | Calculated from final titer | Aim for 1.5-3x increase | Reflects improved process intensity. |
Experimental Protocols
Protocol 3.1: Parallel Shake Flask Screening for Split-PKS Strains
Purpose: To evaluate and select the best-performing split-PKS strain variants under controlled, small-scale conditions prior to bioreactor studies.
Materials:
- Engineered E. coli or S. cerevisiae strains with split-PKS gene constructs.
- Defined production medium (e.g., M9+glucose+appropriate antibiotics).
- Sterile 500 mL baffled shake flasks.
- Temperature-controlled shaker incubator.
- Spectrophotometer for OD measurement.
- Sampling tools (sterile pipettes, cryovials).
Procedure:
- Inoculum Prep: From a frozen glycerol stock, streak strain on an LB+antibiotic plate. Incubate at appropriate temperature (e.g., 30°C) for 24-48h.
- Seed Culture: Pick a single colony into 50 mL of seed medium in a 250 mL flask. Incubate overnight (12-16h) at defined temperature with shaking at 220 rpm.
- Production Culture: Inoculate 50 mL of production medium in a 500 mL baffled flask to an initial OD₆₀₀ of 0.1 from the seed culture. Use biological triplicates.
- Induction: At mid-exponential phase (OD₆₀₀ ~0.6-0.8), induce PKS expression using the defined inducer (e.g., 0.1 mM IPTG for E. coli, or switch to galactose medium for yeast).
- Monitoring: Sample periodically (e.g., every 6-12h) to measure OD₆₀₀, pH (strip), and substrate (e.g., glucose) concentration if possible.
- Harvest: At a defined endpoint (e.g., 72-96h post-induction), take a final sample for OD measurement and product quantification via HPLC-MS.
- Analysis: Calculate growth rate, maximum OD, and final product titer. Select the top 1-2 strains for bioreactor runs based on titer and genetic stability.
Protocol 3.2: Fed-Batch Bioreactor Process for High-Density Cultivation
Purpose: To scale up production of the selected split-PKS strain under controlled, fed-batch conditions to achieve high cell density and maximize product titer.
Materials:
- 5 L bench-top stirred-tank bioreactor with controllers for pH, DO, temperature, and agitation.
- Sterilized vessel with production medium (e.g., 2.5 L initial volume).
- Acid (e.g., 1M H₂SO₄) and base (e.g., 2M NaOH) solutions for pH control in sterilizable bottles.
- Antifoam agent.
- Feed solution: Concentrated carbon/nitrogen source (e.g., 500 g/L glucose + 10 g/L MgSO₄, sterilized by filtration).
- Peristaltic feed pump.
- Exhaust gas analyzer (optional, for CER/OUR calculation).
Procedure: A. Bioreactor Setup & Inoculation:
- Calibrate pH and DO probes prior to sterilization (per manufacturer instructions).
- Add defined initial batch medium to the vessel. Autoclave in-situ or separately.
- Aseptically connect acid/base, antifoam, and feed lines post-sterilization.
- Set process parameters: Temperature = 30°C, initial agitation = 300-400 rpm, aeration = 1.0 vvm (volume of air per volume of liquid per minute), pH = 7.0 (controlled via base addition), DO cascade set to maintain >30% via increasing agitation then pure oxygen.
- Inoculate the bioreactor with a fresh seed culture (from Protocol 3.1, Step 2) to an initial OD₆₀₀ of 0.1.
B. Fed-Batch Operation:
- Batch Phase: Allow cells to grow on the initial medium. Monitor DO, pH, and OD. The DO will drop sharply, indicating active growth.
- Induction: When the initial carbon source is nearly depleted (indicated by a sudden rise in DO), induce PKS expression via a bolus addition of inducer.
- Fed-Batch Phase Initiation: Simultaneously with induction, begin the exponential feed of the concentrated feed solution. The feed rate (F) is calculated to maintain a desired specific growth rate (µ, typically 0.10-0.15 h⁻¹ to balance growth and production) based on the equation: F(t) = (µ/V₀X₀) * exp(µt) / (Yˣ/ˢ * Sf), where V₀ is initial volume, X₀ is initial biomass, Yˣ/ˢ is biomass yield, and Sf is substrate concentration in the feed.
- Process Monitoring: Sample periodically (every 4-8h) for offline analysis: OD₆₀₀, dry cell weight (DCW), substrate concentration, and product titer (HPLC-MS). Record online data (pH, DO, temperature, agitation rate).
- Harvest: Terminate the process at a predefined time (e.g., 24-48h post-induction) or when productivity plateaus. Cool the culture and harvest cells/medium for downstream processing.
Visualization: Experimental Workflow and Metabolic Context
Diagram 1: Scale-up workflow for PKS strains.
Diagram 2: Metabolic flux changes during scale-up.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Scale-Up Experiments
| Item / Reagent | Function & Rationale | Example Product/Specification |
|---|---|---|
| Baffled Shake Flasks | Increases oxygen transfer in shake flask studies by creating turbulence. Essential for meaningful preliminary data. | 500 mL Erlenmeyer flask with 4 baffles, sterile. |
| Defined Production Medium | A chemically defined medium without complex additives (e.g., yeast extract) allows precise control of metabolism and reproducible scale-up. | M9 minimal salts + 20 g/L glucose + trace elements + antibiotics. |
| DO & pH Probes (Sterilizable) | For real-time monitoring and control of the two most critical bioreactor parameters. | Polarographic DO probe, combination pH electrode. |
| Feed Solution (Concentrated) | Enables fed-batch operation to achieve high cell densities and control substrate concentration, preventing overflow metabolism. | 500 g/L Glucose solution, filter-sterilized. |
| Inducer for Heterologous Expression | Precise control of the timing and level of split-PKS gene expression is crucial for balancing growth and production. | Isopropyl β-D-1-thiogalactopyranoside (IPTG) for E. coli systems. |
| Antifoam Emulsion | Controls foam formation in aerated bioreactors, which can interfere with probes and lead to vessel overflow. | Polydimethylsiloxane (PDMS)-based emulsion, sterile. |
| HPLC-MS Standards | For accurate quantification and identification of the target polyketide and potential intermediates in complex broth samples. | Pure analytical standard of the target compound. |
| Rapid Sampling System | Allows aseptic, small-volume sampling from the bioreactor without breaking sterility or disrupting the process. | Sterile, cooled probe with diaphragm valve. |
Application Notes: PKS Gene Splitting in Biosynthesis
Polyketide synthases (PKSs) are modular enzymatic assembly lines responsible for producing diverse polyketide natural products, many of which are clinically valuable. The PKS gene splitting strategy involves dissecting large, contiguous PKS gene clusters into discrete, manageable genetic units. This approach is framed within a broader thesis aiming to overcome host toxicity, expression bottlenecks, and metabolic burden to improve titers and enable the biosynthesis of novel analogues.
Core Advantages:
- Reduced Cellular Complexity & Toxicity: Splitting large PKS pathways into smaller expression units minimizes the metabolic burden on the heterologous host (e.g., E. coli, S. cerevisiae), reducing plasmid instability and toxic intermediate accumulation.
- Enhanced Flexibility & Modularity: Independent transcriptional control of split modules allows for fine-tuning of the stoichiometry of enzyme subunits. This facilitates combinatorial biosynthesis, where modules from different pathways can be mixed and matched to create "unnatural" natural products.
- Improved Protein Folding & Solubility: Expressing large, multi-domain PKS proteins often leads to aggregation and misfolding. Smaller, split subunits are more reliably expressed in soluble, active forms.
Key Trade-offs:
- Increased Operational Complexity: Managing multiple expression vectors, promoters, and cultivation conditions is more labor-intensive than a single-operon approach.
- Potential Yield Penalty: Suboptimal inter-module communication or co-factor channeling between split units can lead to reduced flux through the pathway and lower ultimate yield compared to a perfectly coordinated native megasynthase.
- Scaling Challenges: Laboratory-scale success in shaken flasks does not always translate to scalable fermentation processes due to compounded regulatory and metabolic imbalances.
Quantitative Data Summary:
Table 1: Comparative Performance of Contiguous vs. Split PKS Expression Systems for Erythromycin Precursor (6-DEB) Production in E. coli.
| Expression Strategy | Host Strain | Max Titer (mg/L) | Process Complexity | Flexibility for Engineering | Key Limitation |
|---|---|---|---|---|---|
| Contiguous PKS (DEBS 1-3) | E. coli BAP1 | 15 - 25 | Low | Low | Host toxicity, low soluble protein |
| Split Modules (DEBS 1, 2, 3) | E. coli BL21(DE3) | 70 - 110 | Medium | Medium | Inter-module transfer efficiency |
| Split Modules + Optimized Chassis | E. coli K207-3 | 250 - 300 | High | High | Multi-vector stability at scale |
Table 2: Impact of Promoter Balancing on Yield in a Split Tri-Modular PKS System.
| Promoter Strength Combination (Mod1-Mod2-Mod3) | Relative Protein Expression Ratio | Final Product Titer (Relative %) |
|---|---|---|
| Strong-Strong-Strong | 1.0 : 0.9 : 1.2 | 100% |
| Strong-Medium-Weak | 1.0 : 0.5 : 0.3 | 210% |
| Medium-Strong-Medium | 0.4 : 1.0 : 0.5 | 165% |
Detailed Experimental Protocols
Protocol 1: Golden Gate Assembly for Constructing Split PKS Expression Vectors
Objective: To assemble multiple split PKS modules, each under an inducible promoter, into compatible expression vectors for co-expression.
Materials: See "Scientist's Toolkit" below. Procedure:
- Design & Amplification: Design primers to amplify each PKS module (e.g., DEBS Module 2) from template DNA, adding appropriate Type IIS restriction enzyme overhangs (e.g., BsaI sites).
- Golden Gate Reaction:
- Set up a 20 µL reaction: 50 ng of each PCR-purified module fragment, 50 ng of destination vector (linearized, compatible antibiotic resistance), 1.5 µL T4 DNA Ligase, 1 µL BsaI-HFv2, 2 µL 10x T4 Ligase Buffer.
- Cycle in a thermocycler: 30 cycles of (37°C for 3 min, 16°C for 4 min), then 50°C for 5 min, 80°C for 10 min.
- Transformation: Transform 2 µL of the reaction into competent E. coli DH5α. Plate on LB agar with appropriate antibiotic.
- Screening & Validation: Screen colonies by colony PCR and validate constructs by diagnostic restriction digest and Sanger sequencing.
Protocol 2: Fed-Batch Fermentation for Titer Evaluation of Split PKS Systems
Objective: To evaluate the ultimate yield of a split PKS system under controlled, scalable conditions.
Materials: Bioreactor, defined fermentation media, ammonia hydroxide, feeding solution (50% glycerol, 12% yeast extract), gas mix (O2, N2, air). Procedure:
- Inoculum Prep: Transform all split PKS vectors into the production chassis (e.g., E. coli K207-3). Grow a single colony overnight in 10 mL LB with antibiotics.
- Bioreactor Setup: Inoculate 1L of defined medium in a 2L bioreactor to an initial OD600 of 0.1. Set conditions: 30°C, pH 6.8 (controlled with NH4OH), dissolved oxygen (DO) at 30%.
- Batch Phase: Allow cells to grow until the carbon source is depleted (marked by a sharp rise in DO).
- Induction & Feed Phase: At this point, induce with IPTG (0.5 mM) and begin exponential feeding of the glycerol/yeast extract solution to maintain a specific growth rate (µ) of 0.15 h⁻¹.
- Monitoring & Harvest: Sample periodically to monitor OD600, product titer (via LC-MS), and substrate/byproduct levels. Continue fermentation for 24-36 hours post-induction. Harvest cells by centrifugation for product extraction and quantification.
Visualizations
Diagram 1: PKS Splitting Strategy Logic Flow (86 chars)
Diagram 2: Split PKS Multi-Vector Co-expression Workflow (74 chars)
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for PKS Gene Splitting Experiments
| Item | Function/Description | Example Product/Catalog |
|---|---|---|
| Type IIS Restriction Enzymes | Enable Golden Gate assembly with seamless, scarless ligation of DNA fragments. | BsaI-HF v2, Esp3I (NEB) |
| Modular Expression Vectors | Compatible plasmid set with different antibiotic markers and inducible promoters. | pET Duet series, pCDF Duet (Novagen) |
| Specialized E. coli Chassis | Engineered strains deficient in competitive pathways and enhanced for PKS expression. | E. coli BAP1, K207-3, BL21(DE3)* |
| Inducers for Tunable Control | Small molecules for independent, dose-dependent induction of split modules. | IPTG, Anhydrotetracycline (aTc), L-Arabinose |
| LC-MS/MS System | Critical for quantifying intermediate and final product titers with high sensitivity. | Agilent 6470 Triple Quad, Thermo Q-Exactive |
| Affinity Chromatography Resins | For purification of His- or GST-tagged split PKS subunits for in vitro assays. | Ni-NTA Superflow (Qiagen), GSTrap (Cytiva) |
| Defined Fermentation Media | Chemically defined media for reproducible, high-density fermentation. | M9 Minimal Media, Studier's Autoinduction Media |
Conclusion
The strategic splitting of PKS genes represents a paradigm-shifting engineering solution to the long-standing challenge of heterologously expressing these complex biosynthetic machineries. By deconstructing megaenzymes into functional, co-expressed subunits, researchers can significantly improve protein solubility, reduce cellular burden, and gain unprecedented modular control over pathway architecture. This guide has traversed the journey from foundational principles through practical implementation, troubleshooting, and rigorous validation. The future of this field lies in integrating split-PKS strategies with other synthetic biology tools—such as machine learning for split-site prediction, CRISPR-mediated genome editing for chassis optimization, and automated high-throughput screening. This convergence promises to accelerate the discovery and scalable production of novel polyketide-based therapeutics, opening a new frontier in biomedicine and industrial biotechnology.