Strategies for Overcoming Enzyme Solubility and Expression Challenges in Biopharmaceutical Research
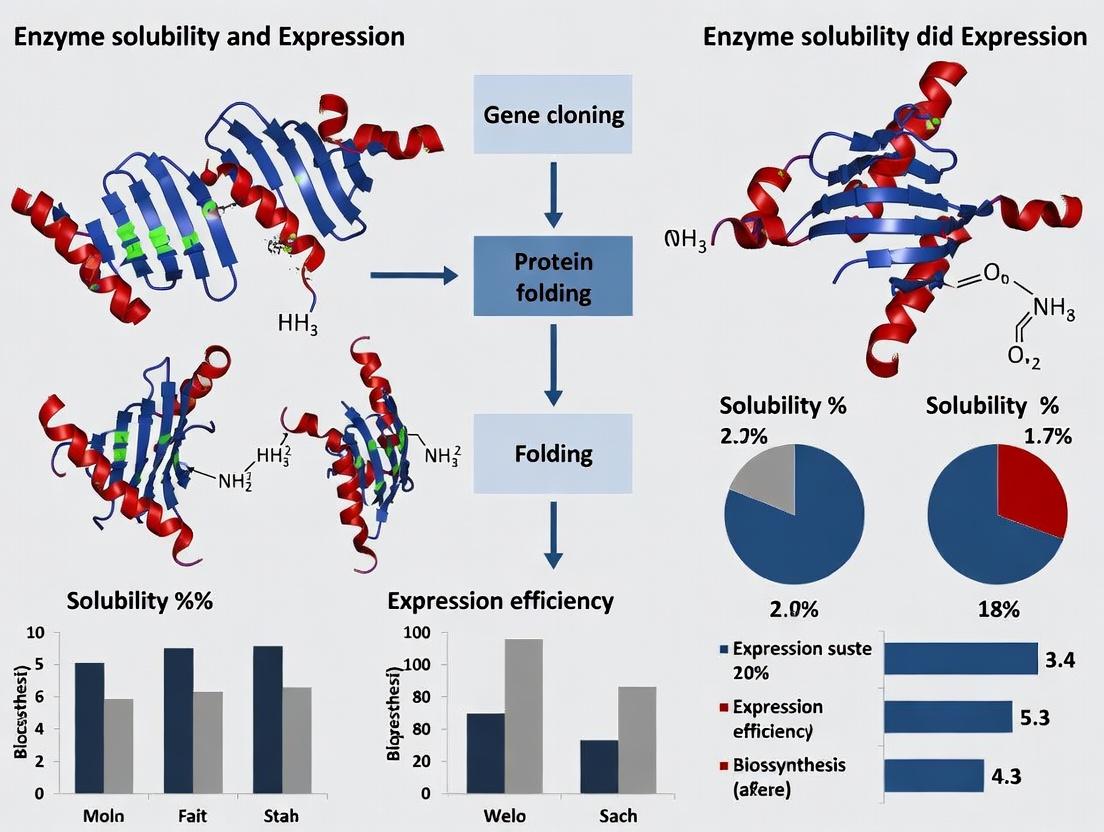
This article provides a comprehensive guide for researchers and drug development professionals facing enzyme solubility and expression issues.
Strategies for Overcoming Enzyme Solubility and Expression Challenges in Biopharmaceutical Research
Abstract
This article provides a comprehensive guide for researchers and drug development professionals facing enzyme solubility and expression issues. It covers foundational understanding of aggregation mechanisms and inclusion body formation, explores methodological approaches like fusion tags and chaperone co-expression, details troubleshooting protocols for optimization, and compares validation techniques. The content synthesizes current best practices to enable efficient production of functional enzymes for therapeutic and diagnostic applications.
Understanding the Root Causes: Why Enzymes Become Insoluble and Fail to Express
Technical Support Center
Troubleshooting Guide: Common Solubility & Expression Issues
Issue Category: Low or No Expression
- Potential Cause: Toxic protein, codon bias, poor transcription/translation initiation.
- Solution: Use a lower-copy plasmid, optimize codons for the host, check promoter strength and RBS, use a lower induction temperature (e.g., 18°C), reduce inducer concentration.
Issue Category: Protein Expressed but Insoluble (Inclusion Bodies)
- Potential Cause: Misfolding due to rapid synthesis, lack of chaperones, hydrophobic protein nature, oxidizing vs. reducing environment mismatch.
- Solution: Reduce expression temperature and rate, co-express with chaperones (GroEL/ES, DnaK/J), test fusion tags (MBP, GST), switch host strain (e.g., Origami for disulfide bonds), screen buffers and solubility enhancers.
Issue Category: Soluble but Non-Functional/Unstable
- Potential Cause: Improper folding, lack of post-translational modifications (PTMs), cofactor absence, proteolysis.
- Solution: Purify using affinity and size-exclusion chromatography, add cofactors during lysis, use protease inhibitors, consider eukaryotic hosts (Sf9, HEK293) for PTMs.
Issue Category: Aggregation During Purification
- Potential Cause: Exposure to non-physiological conditions, concentration too high, loss of cofactors.
- Solution: Optimize pH and salt in buffers, include mild denaturants (e.g., Arg, GdnHCl), use stabilizing ligands, avoid excessive concentration.
Frequently Asked Questions (FAQs)
Q1: My enzyme expresses entirely in inclusion bodies in E. coli. What is my first step to recover soluble protein? A1: The first step is to screen different induction conditions. Lower the temperature to 18-25°C, reduce the inducer concentration (e.g., 0.1 mM IPTG), and shorten induction time. If this fails, consider solubilizing the inclusion bodies with denaturants (6-8 M Urea/GdnHCl) and attempting refolding via dialysis or dilution.
Q2: How do I choose between E. coli strains like BL21(DE3), Rosetta, and Origami for expression? A2: Choose based on your protein's needs:
- BL21(DE3): Standard workhorse for most proteins.
- Rosetta: Supplies rare tRNAs for proteins with codons not optimal for E. coli.
- Origami: Provides a more oxidizing cytoplasm to promote disulfide bond formation.
- ArcticExpress: Co-expresses chaperonins for difficult-to-fold proteins.
Q3: What are the most effective fusion tags for improving solubility, and how do I remove them? A3: Maltose-Binding Protein (MBP) and Glutathione-S-transferase (GST) are highly effective solubility enhancers. SUMO tag often enhances solubility and allows for very efficient cleavage by Ulp1 protease. Tags are typically removed by proteolytic cleavage with site-specific proteases (TEV, Thrombin, Factor Xa, HRV 3C) encoded between the tag and your protein.
Q4: During purification, my protein elutes in the void volume of the size-exclusion column. What does this mean? A4: This typically indicates high-molecular-weight aggregation. Your protein is forming large complexes that cannot enter the resin pores. Immediately check the purification buffer pH and ionic strength, add a mild reducing agent (if applicable), and include 150-500 mM NaCl to reduce non-specific aggregation. Re-evaluate your lysis and initial purification steps for stressors.
Q5: How can I quickly assess if my purified protein is correctly folded and functional? A5: Perform a multi-pronged validation:
- Analytical SEC: A single, symmetric peak at the expected molecular weight.
- Circular Dichroism (CD): Spectrum matching the expected secondary structure (alpha-helix, beta-sheet).
- Intrinsic Fluorescence: Tryptophan emission spectrum peak (~350 nm for exposed, ~330 nm for buried).
- Activity Assay: Use a known biochemical assay to confirm enzymatic function.
Table 1: Common Solubility-Enhancing Fusion Tags Comparison
| Tag | Size (kDa) | Elution Method | Cleavage Protease | Key Advantage |
|---|---|---|---|---|
| MBP | ~42.5 | Maltose | TEV, Factor Xa | Strong solubility enhancer, gentle elution |
| GST | ~26 | Reduced Glutathione | Thrombin, PreScission | Good for dimeric proteins, easy purification |
| SUMO | ~12 | Imidazole (if His-tagged) | Ulp1 | High solubility, efficient & precise cleavage |
| NusA | ~55 | Imidazole (if His-tagged) | TEV | Very effective for difficult prokaryotic proteins |
| Trx | ~12 | Imidazole (if His-tagged) | Enterokinase | Enhances solubility of proteins with disulfides |
Table 2: Troubleshooting Induction Conditions for Solubility
| Condition | Typical Range | Effect on Solubility | Recommended For |
|---|---|---|---|
| Temperature | 16°C - 37°C | Lower temp slows folding, aids solubility | Aggregation-prone proteins |
| IPTG Concentration | 0.01 - 1 mM | Lower conc. reduces expression rate | Toxic proteins, inclusion body formation |
| Induction Time | 2 - 16 hours | Shorter time can reduce aggregation | Rapidly aggregating proteins |
| Host Strain | BL21, Rosetta, etc. | Specialized strains assist folding | Disulfide bonds, codon bias, chaperone need |
Experimental Protocols
Protocol 1: Small-Scale Expression & Solubility Screening
- Transform expression plasmid into appropriate E. coli host strains.
- Inoculate 5 mL cultures (with antibiotic) and grow overnight at 37°C.
- Dilute 1:100 into fresh media (2-5 mL per condition) in a 24-well block.
- Induce at varying conditions (e.g., 18°C/0.1 mM IPTG, 30°C/0.5 mM IPTG) at mid-log phase (OD600 ~0.6).
- Harvest cells 4-18 hours post-induction by centrifugation.
- Lysis: Resuspend pellet in 500 µL lysis buffer (Lysozyme, Benzonase, protease inhibitors). Lyse by sonication or freeze-thaw.
- Separation: Centrifuge at 15,000 x g for 20 min. Separate soluble (supernatant) and insoluble (pellet) fractions.
- Analysis: Analyze both fractions by SDS-PAGE to assess expression level and solubility ratio.
Protocol 2: Refolding from Inclusion Bodies
- Express & Harvest: Induce high-level expression (37°C, 1 mM IPTG) to accumulate inclusion bodies (IBs). Pellet cells.
- Wash: Resuspend cell pellet in IB Wash Buffer (20 mM Tris pH 8.0, 2 M Urea, 1% Triton X-100). Centrifuge. Repeat with buffer without Triton.
- Solubilize: Dissolve IBs in Denaturation Buffer (6-8 M GuHCl or Urea, 20-50 mM Tris, 1-10 mM DTT/TCEP, pH 8-9). Incubate 1 hour at RT with gentle mixing. Centrifuge to clarify.
- Refold: Dilute or dialyze the denatured protein drop-wise into Refolding Buffer (20 mM Tris, 150 mM NaCl, 0.5 M L-Arginine, 2 mM GSH/GSSG redox pair, pH 8.0). Typical 1:10 to 1:100 dilution.
- Concentrate & Purity: Concentrate using centrifugal filters. Purify via SEC or affinity chromatography to isolate correctly folded monomer.
Diagrams
Diagram 1: Solubility Troubleshooting Decision Tree
Diagram 2: Key Host Strains for Soluble Expression
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function in Solubility/Expression Research |
|---|---|
| pET Expression Vectors | High-copy plasmids with strong T7 promoter for controlled, high-level protein expression in E. coli. |
| Chaperone Plasmid Sets | Plasmids co-expressing GroEL/ES or DnaK/J-GrpE to assist in vivo folding of aggregation-prone proteins. |
| Commercial Lysis Buffers | Optimized, ready-to-use buffers containing lysozyme, detergents, and DNase to improve lysis efficiency and sample clarity. |
| Solubility Test Reagents | Kits containing pre-formulated buffers with various pH, salts, and detergents for rapid screening of soluble conditions. |
| Protease Inhibitor Cocktails | Broad-spectrum or specific inhibitors (e.g., PMSF, EDTA, Pepstatin) to prevent proteolytic degradation during purification. |
| TEV Protease | Highly specific, recombinant protease for efficient cleavage of fusion tags with minimal unwanted side-cleavage. |
| L-Arginine Hydrochloride | Common chemical chaperone added to lysis and purification buffers (0.5-1 M) to suppress protein aggregation. |
| HIS-Select Nickel Affinity Gel | High-capacity, nickel-charged resin for robust purification of polyhistidine-tagged proteins under native or denaturing conditions. |
| Size-Exclusion Chromatography Standards | Protein mixtures of known molecular weight for calibrating SEC columns to assess protein oligomerization state. |
Technical Support Center: Troubleshooting Soluble Expression
FAQ 1: Why is my recombinant protein expressed entirely as inclusion bodies? Answer: Inclusion body (IB) formation is a common stress response in heterologous expression, primarily due to the high local concentration of nascent polypeptide chains overwhelming the host cell's folding machinery. Common triggers include:
- High Expression Rate: Excessive T7 or strong promoter activity leads to rapid synthesis.
- Aggregation-Prone Sequences: Inherent low solubility, hydrophobic patches, or missing native partners.
- Non-Physiological Host Environment: Differences in pH, ionic strength, redox potential, and chaperone availability.
- Incorrect Disulfide Bond Formation: In the reducing cytoplasm of E. coli.
FAQ 2: How can I reduce inclusion body formation and promote soluble expression? Answer: Implement a multi-parameter optimization strategy. Refer to the protocol below.
Experimental Protocol: Systematic Optimization for Soluble Expression Title: Screening Parameters for Enhanced Soluble Yield Objective: To identify conditions that maximize soluble expression of a target protein in E. coli. Materials: See "Research Reagent Solutions" table. Procedure:
- Clone & Strain Selection: Clone gene into vectors with different tags (e.g., MBP, GST, Trx) and test expression in various E. coli strains (e.g., BL21(DE3), Origami B, Rosetta-gami).
- Temperature Test: Transform selected vector into strains. For each, inoculate 5 mL cultures, grow to OD600 ~0.6, induce with 0.1-1.0 mM IPTG.
- Split each culture into two: incubate one at 37°C and one at 18°C for 16-20 hours post-induction.
- IPTG Concentration Test: Induce cultures at the better temperature from step 2 with IPTG concentrations of 0.1 mM, 0.5 mM, and 1.0 mM.
- Media & Additive Test: Grow cultures in LB, TB, and Auto-induction media. Supplement selected media with additives (e.g., 1% glucose, 0.5 M sorbitol, 5 mM betaine).
- Harvest & Analysis: Pellet cells. Resuspend in Lysis Buffer. Lyse by sonication. Centrifuge at 15,000 x g for 30 min at 4°C to separate soluble (supernatant) and insoluble (pellet) fractions.
- Analysis: Analyze both fractions by SDS-PAGE. Quantify band intensity to calculate soluble/insoluble ratio.
Summary of Key Optimization Data
| Parameter | Condition Tested | Typical Impact on Solubility (Relative) | Recommended First Test |
|---|---|---|---|
| Expression Temperature | 37°C | Low (Baseline) | Compare 37°C vs. 18°C |
| 18-25°C | High | ||
| Induction OD600 | 0.4-0.6 | Medium | 0.6 |
| >2.0 (Auto-induction) | Medium-High | Use for screening | |
| IPTG Concentration | 1.0 mM | Low | Test 0.1, 0.5, 1.0 mM |
| 0.1-0.5 mM | High | ||
| Host Strain | BL21(DE3) | Baseline | Standard test |
| Origami B (Disulfide) | High for disulfide proteins | For cysteinerich targets | |
| Rosetta (Rare tRNAs) | Medium-High | For codon bias | |
| Fusion Tag | His-tag only | Low (Baseline) | Co-express with tag |
| MBP, GST, Trx | Very High | Test MBP first | |
| Media Additives | None | Baseline | Test Sorbitol/Betaine |
| Osmolytes (e.g., Sorbitol) | Medium |
Research Reagent Solutions: Essential Materials
| Item | Function & Rationale |
|---|---|
| pMAL or pGEX Vectors | Vectors for MBP or GST fusion tags; enhance solubility and provide affinity handles. |
| E. coli Strain: Origami B | Mutant strain with trxB/gor mutations promoting disulfide bond formation in cytoplasm. |
| E. coli Strain: BL21(DE3)pLysS | Provides tight control of basal expression before induction, useful for toxic proteins. |
| Autoinduction Media | Allows high-density growth with automatic induction; useful for screening. |
| Lysozyme & Benzonase | Enzymatic lysis and degradation of genomic DNA to reduce viscosity in lysates. |
| CHAPS or Triton X-100 | Mild detergents used in lysis buffers to aid solubilization of membrane-associated proteins. |
| Protease Inhibitor Cocktail | Prevents degradation of the target protein during cell lysis and purification. |
| Urea & Guanidine HCl | Chaotropic agents for denaturing and solubilizing proteins from inclusion bodies for refolding. |
Diagram 1: Protein Fate in Heterologous Expression
Diagram 2: Solubility Optimization Workflow
Diagram 3: Inclusion Body Refolding Pathway
Technical Support Center: Troubleshooting Enzyme Solubility & Expression
FAQs & Troubleshooting Guides
Q1: My recombinant enzyme is expressed entirely in insoluble inclusion bodies in E. coli. What intrinsic and extrinsic factors should I investigate first?
A: This is a common issue. Follow this systematic troubleshooting guide.
- Intrinsic Factor (Sequence): Check for rare codons for your host (use codon adaptation index calculators). Verify the presence of hydrophobic patches or unstructured regions via in silico analysis. Consider adding solubility-enhancing tags (e.g., MBP, GST, SUMO) to the N- or C-terminus.
- Extrinsic Factor (Host Cell Environment): Reduce the induction temperature (e.g., to 18-25°C). Use a lower concentration of inducer (e.g., 0.1 mM IPTG) to slow protein synthesis and favor proper folding. Co-express chaperone proteins (e.g., GroEL/ES, DnaK/DnaJ/GrpE).
- Extrinsic Factor (Experimental Conditions): Lysis buffer composition is critical. Ensure sufficient ionic strength, pH buffering, and the presence of mild reducing agents (e.g., 1-5 mM DTT).
Q2: I switched from E. coli to a mammalian expression system, but my protein yield is very low. What host-cell extrinsic factors are key?
A: Mammalian systems offer proper folding and PTMs but have lower yields.
- Optimize Transfection & Selection: Use high-quality plasmid DNA and optimize the DNA:transfection reagent ratio. Generate stable polyclonal or clonal cell lines under selective pressure.
- Media & Feed Optimization: Use specialized expression media and consider fed-batch strategies to extend cell viability and protein production.
- Cell Line Choice: Consider alternative lines (HEK293, CHO, insect cells) based on your enzyme's required post-translational modifications.
Q3: How do I experimentally determine if my enzyme's insolubility is due to intrinsic aggregation propensity?
A: Perform the following protocol:
- Bioinformatics Analysis: Use tools like TANGO, AGGRESCAN, or PaRAT to predict aggregation-prone regions (APRs).
- Truncation/Mutation: Design constructs that remove or mutate hydrophobic residues in predicted APRs (e.g., replace Ile, Leu, Val with Lys, Arg, Glu).
- Comparative Solubility Assay: Express and purify both wild-type and mutant proteins under identical extrinsic conditions (host, temperature, lysis buffer).
- Quantify: Compare the soluble fraction yield via SDS-PAGE densitometry (see Table 1).
Q4: My enzyme is soluble but inactive. Could extrinsic experimental conditions during lysis or purification be the cause?
A: Absolutely. Loss of activity often stems from improper buffer conditions or protein handling.
- Buffer Composition: The enzyme may require specific cofactors (Mg²⁺, Zn²⁺, NADH), coenzymes, or stabilizing ligands in the lysis and storage buffers. Check literature for known requirements.
- pH & Redox Environment: A shift from the optimal pH can denature the protein. Ensure correct buffer (e.g., HEPES for pH 7.0-8.0). For enzymes with disulfide bonds, optimize the redox buffer (GSH/GSSG ratio) or use a prokaryotic host with a oxidative periplasm (e.g., E. coli origami strains).
- Purification Speed & Temperature: Always perform purification at 4°C and use rapid protocols to minimize protease activity and denaturation.
Data Presentation
Table 1: Impact of Extrinsic Factors on Soluble Yield of Model Enzyme (Galactosidase) in E. coli
| Extrinsic Factor | Condition A | Condition B | Condition C | Soluble Yield (% of Total) |
|---|---|---|---|---|
| Induction Temperature | 37°C | 25°C | 18°C | 15% vs. 60% vs. 75% |
| IPTG Concentration | 1.0 mM | 0.5 mM | 0.1 mM | 20% vs. 45% vs. 70% |
| Host Strain | BL21(DE3) | C41(DE3) | Rosetta-gami 2 | 30% vs. 55% vs. 80% |
| Lysis Buffer Additive | None | 0.5 M Arg / 0.5 M GndHCl | 1% N-Lauroylsarcosine | 40% vs. 65% vs. 10% |
Table 2: Key Research Reagent Solutions for Solubility & Expression Troubleshooting
| Reagent / Material | Primary Function in Troubleshooting |
|---|---|
| Solubility-Tag Vectors (pMAL, pGEX, pET-SUMO) | Fusion tags (MBP, GST, SUMO) enhance solubility and provide an affinity handle for purification. |
| Chaperone Plasmid Kits (Takara, Agilent) | Co-expression plasmids for GroEL/ES or DnaK/J/E to assist in vivo folding. |
| E. coli Strains: Origami, Rosetta, C41/C43 | Provide disulfide bond formation (Origami), rare tRNA (Rosetta), or reduced toxicity (C41/43). |
| Detergents & Additives: CHAPS, DDM, Arg, GndHCl | Aid in solubilizing proteins from membranes or inclusion bodies during lysis. |
| Protease Inhibitor Cocktails (e.g., PMSF, Pepstatin, E-64) | Prevent proteolytic degradation during cell lysis and purification. |
| Affinity Chromatography Resins (Ni-NTA, Glutathione, Amylose) | For rapid capture and purification of His-, GST-, or MBP-tagged fusion proteins. |
| Size-Exclusion Chromatography (SEC) Column | Final polishing step to remove aggregates and buffer exchange into storage conditions. |
Experimental Protocols
Protocol: High-Throughput Screening of Expression Conditions Objective: Identify optimal extrinsic conditions (temperature, inducer concentration, host strain) for soluble expression.
- Clone gene of interest into expression vector with affinity tag.
- Transform a panel of E. coli expression strains (BL21(DE3), C43(DE3), Rosetta2).
- Inoculate deep-well plates with 1 mL auto-induction media per well.
- Induce with a gradient of IPTG (0.01, 0.1, 0.5, 1.0 mM) across columns.
- Incubate with shaking at a gradient of temperatures (18°C, 25°C, 30°C, 37°C) across rows.
- Harvest cells by centrifugation after 18-24 hours.
- Lysis via chemical/ enzymatic (lysozyme) method in 96-well format.
- Clarify lysates by centrifugation. Save soluble (supernatant) and insoluble (pellet) fractions.
- Analyze fractions by SDS-PAGE (e.g., using precast gels) to determine soluble yield under each condition.
Protocol: Refolding from Inclusion Bodies
- Express & Pellet: Express protein at 37°C to drive to inclusion bodies (IBs). Harvest cells.
- Wash IBs: Resuspend pellet in IB Wash Buffer (20 mM Tris-HCl pH 7.5, 2 M Urea, 1% Triton X-100). Centrifuge. Repeat with buffer without Triton.
- Solubilize: Dissolve IB pellet in Denaturing Buffer (6 M GdnHCl, 20 mM Tris pH 8.0, 10 mM DTT) for 1 hour at room temperature.
- Clarify: Centrifuge to remove any insoluble debris.
- Refold: Rapidly dilute the denatured protein 50-fold into chilled Refolding Buffer (20 mM Tris pH 8.0, 0.5 M L-Arg, 2 mM reduced glutathione, 0.2 mM oxidized glutathione) with gentle stirring. Let stand at 4°C for 12-36 hours.
- Concentrate & Purity: Concentrate refolded protein using centrifugal filters, then purify by SEC to isolate monomeric, correctly folded species.
Visualizations
Title: Systematic Troubleshooting Workflow for Enzyme Solubility
Title: Interplay of Intrinsic & Extrinsic Factors on Expression Outcome
Troubleshooting Guide: Enzyme Solubility & Expression Issues
Q1: My expressed recombinant enzyme is entirely insoluble. What are the first steps to troubleshoot?
A: Insolubility often indicates protein misfolding or aggregation. Follow this systematic protocol:
- Confirm the Issue: Centrifuge the lysate at 20,000 x g for 30 min at 4°C. Analyze supernatant (soluble) and pellet (insoluble) fractions by SDS-PAGE.
- Lower Expression Temperature: Reduce the growth temperature to 18-25°C post-induction to slow protein synthesis and improve folding.
- Optimize Induction: Use a lower inducer concentration (e.g., 0.1-0.5 mM IPTG) and shorter induction times (2-4 hours).
- Co-express Molecular Chaperones: Use E. coli strains like BL21(DE3)pLysS Rosetta2 or co-transform with plasmids expressing GroEL/GroES.
- Screen Solubility-Enhancing Tags: Test N- or C-terminal fusion tags (e.g., MBP, GST, SUMO, Trx) using a modular vector system.
Q2: My enzyme is soluble but shows no catalytic activity. How do I diagnose lost activity?
A: Soluble but inactive protein suggests improper folding, missing cofactors, or post-translational modifications.
Diagnostic Workflow:
- Check for Inclusion Bodies in Soluble Fraction: Perform native PAGE or dynamic light scattering to detect soluble aggregates.
- Verify Cofactor Incorporation: Supplement purification and assay buffers with essential cofactors (e.g., Mg²⁺, Zn²⁺, NADH, heme). See Table 1 for common requirements.
- Assess Oligomeric State: Use size-exclusion chromatography (SEC) or analytical ultracentrifugation to determine if the protein forms the correct multimer.
- Perform a Refolding Screen: If aggregates are present, denature the protein in 6 M Guanidine-HCl and refold using a commercial screen (e.g., Hampton Research QuickFold).
Table 1: Common Enzyme Cofactors & Diagnostic Additives
| Cofactor/Additive | Typical Concentration | Function | Example Enzyme Class |
|---|---|---|---|
| Mg²⁺ | 1-10 mM | Catalytic metal ion | Kinases, Polymerases |
| Zn²⁺ | 10-100 µM | Structural/Catalytic metal ion | Metalloproteases, Dehydrogenases |
| DTT / TCEP | 1-5 mM | Reduces disulfide bonds, prevents oxidation | Cysteine-dependent enzymes |
| PLP (Pyridoxal Phosphate) | 50-200 µM | Catalytic coenzyme | Aminotransferases, Decarboxylases |
| NAD(P)H | 100-500 µM | Redox cofactor | Oxidoreductases, Reductases |
Q3: How do solubility issues directly lead to altered pharmacokinetics (PK) in drug development?
A: Poor enzyme solubility during in vitro testing creates misleading data that fails to predict in vivo behavior, leading to PK failures.
- False Negative Activity: An insoluble but otherwise functional enzyme target will show low activity in HTS, causing promising drug candidates to be discarded.
- Altered Binding Kinetics: Aggregated enzymes can non-specifically bind compounds, leading to inaccurate IC₅₀/Kᵢ measurements.
- Incorrect SAR: Structure-activity relationships (SAR) are built on faulty activity data, guiding medicinal chemistry in the wrong direction.
- Downstream PK Impact: A compound optimized against an insoluble/misfolded enzyme may have completely different binding and off-target profiles in vivo, resulting in unexpected clearance, volume of distribution, or toxicity.
Protocol: Assessing Compound Aggregation in Enzyme Assays To rule out false positives/negatives due to compound or enzyme aggregation:
- Test with Detergent: Include 0.01% Triton X-100 in the activity assay. A significant change in inhibition suggests compound aggregation.
- Perform Dynamic Light Scattering (DLS): Measure the enzyme preparation before assay. A polydisperse sample indicates aggregates.
- Use a Counter-Screen: Run a non-enzymatic assay (e.g., fluorescence interference, redox activity) to identify promiscuous inhibitors.
Q4: What strategies can rescue activity for a poorly soluble pharmacokinetic enzyme target?
A: Implement a combination of expression, purification, and formulation strategies.
Detailed Protocol: Solubilization & Refolding for PK Studies Materials: Lysis Buffer (50 mM Tris, 300 mM NaCl, pH 8.0), Wash Buffer (Lysis Buffer + 1% Triton X-100), Denaturation Buffer (50 mM Tris, 8 M Urea, 10 mM DTT, pH 8.0), Refolding Buffer (50 mM Tris, 150 mM NaCl, 2 mM Reduced Glutathione, 0.2 mM Oxidized Glutathione, 10% Glycerol, pH 8.0).
- Isolate Inclusion Bodies: Resuspend cell pellet in Lysis Buffer. Lyse by sonication. Centrifuge at 20,000 x g for 30 min. Wash pellet twice with Wash Buffer.
- Solubilize: Solubilize pellet in Denaturation Buffer for 1 hour with gentle stirring.
- Refold by Rapid Dilution: Clarify the denatured solution by centrifugation. Rapidly dilute the supernatant 1:50 into cold Refolding Buffer. Stir gently for 12-16 hours at 4°C.
- Concentrate & Purify: Concentrate the refolded protein using a centrifugal concentrator (10 kDa MWCO). Purify via SEC in final assay buffer.
- Validate: Confirm monodispersity by SEC-MALS and measure catalytic activity (kₐₜₜ/Kₘ) against a standard substrate.
Frequently Asked Questions (FAQs)
Q: Which solubility tag should I use for my kinase enzyme?
A: Maltose-Binding Protein (MBP) and Glutathione-S-transferase (GST) are highly effective for kinases. MBP often provides superior solubility and can be removed with precise TEV protease cleavage, leaving no artifact residues.
Q: How does buffer composition affect long-term enzyme stability for PK assays?
A: Critical factors include pH (use 20-50 mM buffer capacity), salt type/strength (150-200 mM NaCl/KCl), reducing agents (1 mM TCEP), stabilizers (10% glycerol, 0.5 mg/mL BSA), and non-ionic detergents (0.01% Tween-20). Always avoid repeated freeze-thaw cycles; flash-freeze in single-use aliquots.
Q: My cytochrome P450 enzyme is soluble but loses heme. How can I recover it?
A: This indicates heme misincorporation or loss. Purify the apo-enzyme and perform in vitro heme reconstitution: Incubate the protein with a 2:1 molar ratio of hemin (from a fresh 10 mM stock in DMSO) in buffer containing 5 mM DTT for 1 hour on ice. Remove excess heme via desalting column. Monitor the Soret peak at 450 nm (reduced CO-bound form) for activity.
Research Reagent Solutions Toolkit
| Reagent / Material | Supplier Examples | Function in Solubility/Activity Rescue |
|---|---|---|
| pET-28a(+) Vector | Novagen, MilliporeSigma | T7 expression vector with optional N-/C-terminal His₆-tag for purification. |
| Rosetta2(DE3) E. coli Cells | MilliporeSigma | Supplies rare tRNAs for codons underrepresented in E. coli (e.g., AGG, AGA, AUA). |
| Chaperone Plasmid Set (GroEL/ES, DnaK/J-GrpE) | Takara Bio | Co-expression plasmids to assist proper folding of eukaryotic or complex proteins. |
| Hampton Research QuickFold Kit | Hampton Research | 96-condition screen for optimizing refolding buffers. |
| TEV Protease | Thermo Fisher, homemade | Highly specific protease to remove affinity tags without leaving extra residues. |
| Size-Exclusion Chromatography Columns (Superdex 75/200) | Cytiva | Critical for separating monodisperse, active enzyme from aggregates. |
| TCEP-HCl (Tris(2-carboxyethyl)phosphine) | Gold Biotechnology | Stable, odorless reducing agent to maintain cysteines in reduced state. |
| Protein Stability & Aggregation Dyes (SYPRO Orange, ANS) | Thermo Fisher | Used in thermal shift assays to monitor folding state and identify stabilizing conditions. |
Experimental & Conceptual Diagrams
Title: Downstream Impact of Solubility Issues on Drug Development
Title: Soluble but Inactive Enzyme Troubleshooting Guide
Proven Strategies and Expression Systems for Soluble Enzyme Production
Technical Support Center: Troubleshooting & FAQs
Q1: My recombinant protein, fused with an MBP tag, is expressed in E. coli but remains entirely in the insoluble fraction. What are my primary troubleshooting steps? A: This is a common issue. Follow this systematic protocol:
- Reduce Expression: Lower the induction temperature (to 16-25°C) and reduce IPTG concentration (to 0.1-0.5 mM) or induction time. This slows protein synthesis, allowing proper folding.
- Strain Selection: Use E. coli strains engineered for disulfide bond formation (e.g., SHuffle) if applicable, or chaperone-enriched strains (e.g., ArcticExpress).
- Lysis Buffer Optimization: Supplement lysis buffer with non-denaturing agents (e.g., 0.5-1% Triton X-100, 1-2 M Urea, or 0.5 M Arginine) to weaken hydrophobic interactions.
- Co-express with Chaperones: Co-transform with a plasmid expressing GroEL/ES or DnaK/DnaJ/GrpE chaperone systems.
- Tag Position: If possible, clone the fusion tag (MBP) at the C-terminus instead of the N-terminus, or vice versa.
- Final Resort - Refolding: Solubilize the inclusion bodies in 8 M urea or 6 M guanidine HCl, then refold by dialysis or dilution in the presence of additives like arginine, glycerol, and redox couples.
Q2: I successfully purified my GST-tagged protein via glutathione affinity, but the thrombin cleavage to remove the tag is inefficient (<50%). What factors should I check? A: Inefficient cleavage often stems from suboptimal reaction conditions.
- Verify the Cleavage Site: Ensure the protease recognition sequence is accessible and not sterically hindered. Add a short linker (e.g., GGSGG) between the GST tag and the cleavage site.
- Optimize Cleavage Conditions:
- Enzyme:Substrate Ratio: Titrate from 1:100 to 1:10 (w/w).
- Buffer: Thrombin requires a mild reducing environment. Ensure 1-2 mM DTT or β-mercaptoethanol is present. Use Tris or PBS buffer at pH 7.0-8.5.
- Temperature & Time: Perform cleavage at 4°C for 16-20 hours instead of room temperature for 2-4 hours to reduce non-specific proteolysis.
- Remove Chelators: Ensure no EDTA is present in your buffer, as thrombin requires Ca²⁺.
- Cleavage While Immobilized: Perform the cleavage step while the protein is still bound to the glutathione resin. This often improves accessibility.
Q3: After His-tag purification using IMAC, my protein sample is contaminated with host E. coli proteins. How can I improve purity? A: Contamination is typically due to non-specific binding. Implement these fixes:
Table 1: Troubleshooting IMAC Purity Issues
| Issue & Likely Cause | Recommended Solution | Protocol Adjustment |
|---|---|---|
| Imitazole Carryover | Increase imidazole in wash buffer. | Perform a stepped wash: 20 mM, then 40-50 mM imidazole in binding buffer before elution. |
| Metal Leakage & Weak Binders | Use more stringent wash conditions. | Add 0.5-1 M NaCl and/or 1-5% glycerol to wash buffer to reduce ionic/hydrophobic interactions. |
| Non-specific Binding of Metalloproteins | Include a chelating agent in wash. | Add 1-5 mM MgCl₂ or CaCl₂ to wash buffer to occupy non-specific metal-binding sites on host proteins. |
| Protein Degradation | Use protease inhibitors. | Include a cocktail (e.g., PMSF, leupeptin, pepstatin) in all lysis and purification buffers. Keep samples cold. |
| Tag Inaccessibility | Optimize binding conditions. | Increase salt (300-500 mM NaCl) and detergent (0.05% Tween-20) in binding/wash buffers. |
Q4: My SUMO-tagged protein cleaves efficiently with SUMO protease, but my target protein instantly precipitates after tag removal. How can I prevent this? A: This indicates the SUMO tag was crucial for solubility. The protocol must maintain solubility post-cleavage.
- Cleavage in Presence of Stabilizers: Perform the SUMO protease reaction in a buffer containing solubility enhancers: 0.5-1 M arginine, 10-20% glycerol, 0.01-0.1% CHAPS, or mild denaturants (0.5-1 M urea).
- Rapid Downstream Processing: Immediately load the cleavage reaction onto an ion-exchange or size-exclusion column to separate the target protein from the cleaved tag and protease. The change in buffer environment can help.
- Test Alternative Tags: If the target protein is inherently insoluble, consider switching to a different solubility-enhancing tag (MBP, NusA) that may be left on for downstream activity assays, or use a tandem tag system (e.g., SUMO-His).
Q5: What are the key quantitative differences between common fusion tags that influence choice for enzyme expression? A: Selection is based on yield, solubility enhancement, and ease of removal.
Table 2: Comparative Analysis of Common Fusion Tags
| Tag | Approx. Size (kDa) | Primary Function | Typical Yield (E. coli) | Key Advantage | Key Disadvantage |
|---|---|---|---|---|---|
| His-tag | ~0.8 | Affinity Purification | 1-20 mg/L | Fast, generic, mild elution | Low purity; may not enhance solubility. |
| GST | ~26 | Affinity Purification & Dimerization | 2-10 mg/L | High purity; can aid folding. | Large size may affect activity; elution with glutathione can be harsh. |
| MBP | ~42.5 | Solubility Enhancement | 5-50 mg/L | Potent solubility enhancer. | Very large; may require removal for activity studies. |
| SUMO | ~11 | Solubility & Cleavage | 2-20 mg/L | Enhances solubility/expression; precise, efficient cleavage. | Protease is expensive; cleavage can expose insolubility. |
| Trx | ~12 | Solubility Enhancement (Reducing Env.) | 5-30 mg/L | Good for disulfide-bonded proteins. | Less effective than MBP for severely aggregating proteins. |
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Rationale |
|---|---|
| pMAL / pETM Series Vectors | Cloning vectors for in-frame fusion to Maltose-Binding Protein (MBP), offering different protease cleavage sites. |
| GSH Sepharose / Glutathione Agarose | Affinity resin for one-step purification of GST-tagged proteins via stable thioester bond formation. |
| Ni-NTA / Co²⁺ IMAC Resin | Immobilized metal affinity chromatography resins for purifying polyhistidine-tagged proteins. Cobalt resin offers higher specificity than nickel. |
| SUMO Protease / Ulp1 | Highly specific protease that cleaves after the C-terminal Gly-Gly of the SUMO tag, leaving no residual amino acids on the target. |
| TEV Protease | Highly specific protease (recognizes Glu-Asn-Leu-Tyr-Phe-Gln-Gly) used for cleaving tags, often in tandem with His-tags for purification. |
| Chaperone Plasmid Sets (e.g., pG-KJE8) | Plasmids for co-expressing E. coli chaperone systems (DnaK/DnaJ/GrpE, GroEL/ES) to aid folding of difficult proteins. |
| BL21(DE3) Derivative Strains (Rosetta, SHuffle, ArcticExpress) | Specialized expression hosts providing tRNA for rare codons, disulfide bond formation in the cytoplasm, or cold-adapted chaperones. |
| Solubility Enhancers (L-Arginine, Glycerol, Betaine) | Additives in lysis or cleavage buffers that stabilize proteins, reduce aggregation, and weaken hydrophobic interactions. |
Experimental Protocol: Evaluating Fusion Tag Impact on Soluble Expression
Title: Systematic Screen for Optimal Fusion Tag Objective: To identify the fusion tag (His, MBP, GST, SUMO) that yields the highest amount of soluble, functional enzyme. Method:
- Cloning: Clone the target enzyme sequence into a set of isexpression vectors, each containing a different N-terminal fusion tag (e.g., pET28a-His, pMAL-MBP, pGEX-GST, pE-SUMO). Verify sequences.
- Small-Scale Expression: Transform each construct into an appropriate E. coli expression strain (e.g., BL21(DE3)). Inoculate 5 mL cultures in deep-well plates. Grow at 37°C to OD600 ~0.6. Induce with 0.5 mM IPTG.
- Condition Testing: Divide each culture. Express proteins at two temperatures: 37°C for 4 hours and 18°C for 16-20 hours.
- Soluble Fraction Analysis: a. Harvest cells by centrifugation. Lyse with BugBuster reagent or lysozyme/sonication. b. Centrifuge at 15,000 x g for 20 min to separate soluble (supernatant) and insoluble (pellet) fractions. c. Analyze equal proportions of total, soluble, and insoluble fractions by SDS-PAGE.
- Quantification & Activity: Use densitometry on SDS-PAGE gels to estimate the percentage of soluble protein. For constructs showing high solubility, perform a small-scale affinity purification (appropriate to the tag) and assay for specific enzyme activity.
Visualization: Experimental & Conceptual Diagrams
Title: Workflow for Fusion Tag Solubility Screening
Title: Decision Pathway for Addressing Protein Solubility Issues
Technical Support Center: Troubleshooting Enzyme Solubility & Expression
Troubleshooting Guides
Issue 1: Low or No Protein Expression in E. coli
- Potential Cause: Codon bias, toxic protein, poor plasmid stability, or incorrect induction.
- Solution: Use a strain with rare tRNA plasmids (e.g., BL21-CodonPlus). Test lower induction temperatures (e.g., 16-25°C) and different inducer concentrations. Check plasmid integrity via diagnostic digest.
- Experimental Protocol (IPTG Titration):
- Transform expression plasmid into expression host (e.g., BL21(DE3)).
- Inoculate 5 mL cultures and grow to mid-log phase (OD600 ~0.6).
- Add IPTG to final concentrations of 0.1, 0.5, 1.0 mM.
- Incubate at 16°C, 25°C, and 37°C for 4-18 hours.
- Pellet cells, lyse, and analyze supernatant and pellet by SDS-PAGE.
Issue 2: Protein Insolubility (Inclusion Bodies) in E. coli
- Potential Cause: Aggregation due to rapid expression, lack of chaperones, or reducing environment.
- Solution: Use strains with chaperone plasmids (e.g., BL21(DE3) pG-KJE8). Lower growth temperature post-induction (16-25°C). Consider co-expression with fusion tags (e.g., MBP, SUMO). Screen solubility in lysis buffers with varying pH and salt.
- Experimental Protocol (Solubility Screen):
- Induce expression at low temperature (18°C, overnight).
- Lyse cells in Buffer A (50 mM Tris, 300 mM NaCl, pH 8.0, 1 mg/mL lysozyme).
- Centrifuge at 20,000 x g for 30 min to separate soluble (supernatant) and insoluble (pellet) fractions.
- Resuspend pellet in Buffer A with 6M Urea or Guanidine-HCl.
- Analyze equal % volumes of total, soluble, and insoluble fractions by SDS-PAGE.
Issue 3: Inefficient Secretion in Yeast (Pichia pastoris)
- Potential Cause: Incorrect signal peptide, hyperglycosylation, or ER stress.
- Solution: Test alternative signal peptides (α-MF, SUC2). Use a strain with reduced glycosylation (e.g., pichia pastoris SuperMan5). Optimize methanol induction timeline and concentration.
- Experimental Protocol (Methanol Induction Time Course):
- Grow culture in glycerol medium to high density (OD600 ~10-20).
- Centrifuge and resuspend in methanol-containing medium (0.5-1% v/v).
- Maintain induction for 96 hours, feeding methanol to 0.5% daily.
- Take samples every 24 hours. Centrifuge to separate cells from supernatant.
- Analyze culture supernatant via SDS-PAGE and Western blot.
Issue 4: Low Baculovirus Titer in Insect Cell Systems
- Potential Cause: Poor cell health, suboptimal Multiplicity of Infection (MOI), or incorrect passage of viral stock.
- Solution: Always use cells in mid-log phase (viability >97%). Amplify virus in a stepwise manner (P0 -> P1 -> P2). Determine optimal MOI (typically 0.05-0.1 for amplification, 3-5 for expression) via plaque assay.
- Experimental Protocol (Viral Amplification - Sf9 cells):
- Seed 50 mL of Sf9 cells at 0.5 x 10^6 cells/mL in a 125 mL flask.
- Add P0 viral stock at an MOI of 0.05-0.1.
- Incubate at 27°C, 110 rpm for 72-96 hours.
- Monitor cell viability and granulation. Harvest supernatant when viability drops to ~50% (P1 stock).
- Clarify supernatant by centrifugation (500 x g, 5 min). Store at 4°C protected from light.
Issue 5: Low Transfection Efficiency in Mammalian Cells (HEK293)
- Potential Cause: Poor quality DNA, subconfluent cells, or inefficient transfection reagent.
- Solution: Use endotoxin-free plasmid prep. Transfect when cells are 70-90% confluent. Optimize DNA:reagent ratio using a GFP reporter plasmid.
- Experimental Protocol (PEI-mediated Transfection for HEK293F):
- Grow suspension HEK293F cells to 1.0-1.2 x 10^6 cells/mL, viability >95%.
- For 1L culture, mix 1 mg plasmid DNA with 50 mL fresh medium.
- Add 3 mg linear PEI (pH 7.0) to 50 mL fresh medium.
- Combine DNA and PEI solutions, vortex immediately, incubate 15-20 min at RT.
- Add dropwise to culture. Harvest 48-72 hours post-transfection.
FAQs
Q: Which E. coli strain should I choose for my difficult-to-express enzyme? A: For cytoplasmic expression, start with BL21(DE3) for its protease deficiency. For disulfide-bonded proteins, use SHuffle strains. For codon-optimization, use BL21(DE3) pRARE2. For membrane proteins, consider C41(DE3) or C43(DE3).
Q: How do I choose between Pichia pastoris and Saccharomyces cerevisiae for yeast expression? A: Use this table for a quantitative comparison:
| Feature | Pichia pastoris | Saccharomyces cerevisiae |
|---|---|---|
| Expression Level | Very High (g/L) | Moderate (mg/L) |
| Glycosylation Type | High-mannose, shorter (Man8-14) | Hyper-mannose (Man50-150) |
| Common Plasmid | pPICZ series (AOX1 promoter) | pYES2 (GAL1 promoter) |
| Typical Yield | 1-10 g/L extracellular | 10-100 mg/L intracellular |
| Induction Method | Methanol | Galactose |
Q: What are the key differences between insect cell systems (Sf9 vs. Sf21 vs. High Five) for enzyme production? A:
| Cell Line | Typical Virus | Growth Rate | Max Cell Density | Protein Yield | Notes |
|---|---|---|---|---|---|
| Sf9 | AcMNPV | Moderate | 5-6 x 10^6/mL | High | Robust, standard for amplification |
| Sf21 | AcMNPV | Faster | 4-5 x 10^6/mL | Moderate | More sensitive, used for plaque assays |
| High Five | BmNPV | Fast | 3-4 x 10^6/mL | Very High | Often higher secretion, but less standard |
Q: When should I use transient vs. stable transfection in mammalian cells? A: Use transient transfection (e.g., HEK293T, Expi293F) for rapid protein production (mg-scale in 1 week) and screening. Use stable transfection (e.g., CHO-K1, Flp-In systems) for long-term, consistent production (g-scale over months), requiring antibiotic selection and clonal isolation.
Visualizations
Diagram 1: Host System Decision Pathway
Title: Host Selection for Enzyme Expression
Diagram 2: E. coli Solubility Troubleshooting Workflow
Title: E. coli Solubility Problem-Solving Flow
The Scientist's Toolkit: Key Reagent Solutions
| Reagent / Material | Primary Function | Key Considerations for Enzyme Solubility |
|---|---|---|
| BL21(DE3) E. coli Cells | Standard protein expression host. | Deficient in Lon and OmpT proteases; use for initial trials. |
| SHuffle T7 E. coli Cells | Expression of disulfide-bonded proteins in cytoplasm. | Provides oxidizing environment; essential for cytosolic folding of enzymes with Cys bridges. |
| pET Expression Vectors | High-level, inducible expression in E. coli. | Strong T7 promoter; N- or C-terminal tags (His, GST, MBP) can aid solubility. |
| Linear Polyethylenimine (PEI) Max | High-efficiency transfection of mammalian cells. | Cost-effective for large-scale transient transfections in HEK293 cells. |
| Sf900 III SFM | Serum-free medium for insect cell culture. | Supports high-density growth of Sf9/Sf21 cells for baculovirus expression. |
| PichiaPink Secretion Signal Set | Kit of 4 different secretion signals for Pichia. | Enables rapid screening for optimal enzyme secretion in yeast. |
| HALT Protease Inhibitor Cocktail | Broad-spectrum protease inhibition. | Add to lysis buffer immediately to prevent degradation of soluble enzyme. |
| Detergent Screen Kit (e.g., from Hampton Research) | Set of mild detergents for membrane protein solubilization. | Critical for extracting and stabilizing integral membrane enzymes. |
| Cycloheximide | Eukaryotic translation inhibitor. | Used in pulse-chase experiments to analyze enzyme stability and turnover. |
Technical Support Center: Troubleshooting Guides & FAQs
Frequently Asked Questions (FAQs)
Q1: My target protein is consistently expressed as insoluble aggregates when expressed in E. coli. Which chaperone system should I co-express first? A: Begin with Trigger Factor (TF) and DnaK (Hsp70) co-expression, as they act early on nascent chains. If insoluble aggregates persist, add the GroEL/ES system, which refolds proteins post-translationally. For large, multi-domain proteins (>60 kDa), prioritize GroEL/ES from the start.
Q2: How do I determine if my protein is aggregation-prone before starting experiments? A: Use in-silico prediction tools (e.g., TANGO, AGGRESCAN) to analyze the amino acid sequence for hydrophobic stretches and low-complexity regions. Aggregation propensity scores above defined thresholds indicate a high risk.
Q3: Co-expression with chaperones did not improve solubility. What are the next steps? A: Troubleshoot using this hierarchy:
- Verify Chaperone Expression: Run an SDS-PAGE gel to confirm chaperone overexpression.
- Optimize Conditions: Lower induction temperature (e.g., 18-25°C), reduce inducer concentration (e.g., 0.1 mM IPTG), and use richer growth media.
- Try Combinations: Use plasmids encoding chaperone teams (e.g., pG-KJE8 for DnaK/DnaJ/GrpE and GroEL/ES).
- Consider Fusion Tags: Switch to or add a solubility-enhancing tag (e.g., MBP, SUMO) in tandem with chaperone co-expression.
Q4: What is the key difference between the roles of DnaK and GroEL? A: DnaK (with DnaJ and GrpE) binds to exposed hydrophobic patches on nascent or unfolded polypeptides, preventing aggregation and facilitating folding. GroEL/ES provides an isolated, hydrophilic chamber for single protein molecules to fold unimpaired by crowding or aggregation.
Q5: Can I co-express all major chaperone systems simultaneously? A: Yes, but with caution. While plasmids like pG-Tf2 (TF + GroEL/ES) and pG-KJE8 (DnaK/DnaJ/GrpE + GroEL/ES) exist, overloading the cell with chaperone expression plasmids can cause metabolic burden, reduce target protein yield, and complicate genetic stability.
Troubleshooting Guide
| Symptom | Possible Cause | Recommended Solution |
|---|---|---|
| Low yield of both chaperone and target protein. | Metabolic burden from multiple plasmids/inducers. | Use compatible plasmids with different antibiotic resistance and replication origins. Titrate inducer concentrations (e.g., 0.01-0.5 mM IPTG for trc/lac promoters). |
| Protein is soluble but inactive. | Improper folding despite solubility. | Co-express with GroEL/ES specifically; refine lysis/buffer conditions (add Mg-ATP for chaperone activity in vitro). |
| Chaperone co-expression has no effect. | Chaperones not expressed or inactive. | Check plasmid sequences and induction protocols. For GroEL/ES, ensure Mg2+ and ATP are present in the lysis buffer. |
| Increased cell death or slow growth. | Toxicity of target protein or chaperone overexpression. | Use tightly regulated promoters (e.g., pBAD for arabinose-controlled expression), lower temperature, and use lower-copy number chaperone plasmids. |
Table 1: Chaperone System Characteristics and Success Rates
| Chaperone System | Typical Solubility Increase* | Optimal Target Protein Size/Type | Common Expression Plasmid(s) | Key Cofactors Required |
|---|---|---|---|---|
| Trigger Factor (TF) | 1.5 - 3 fold | Small to medium (<50 kDa), nascent chains | pTf16, pG-Tf2 | None |
| DnaK-DnaJ-GrpE | 2 - 5 fold | Aggregation-prone, hydrophobic domains | pKJE7, pG-KJE8 | ATP, K+ |
| GroEL-GroES | 3 - 10 fold | Large, multi-domain (>60 kDa) | pGro7, pG-Tf2, pG-KJE8 | ATP, Mg2+ |
| Combination (DnaK+GroEL) | 5 - >10 fold | Highly complex, recalcitrant proteins | pG-KJE8 | ATP, Mg2+, K+ |
*Reported fold-increase in soluble fraction varies widely by target protein.
Table 2: Standard Induction Conditions for Chaperone Plasmids
| Plasmid | Chaperones Expressed | Antibiotic | Inducer | Typical Concentration |
|---|---|---|---|---|
| pKJE7 | DnaK, DnaJ, GrpE | Chloramphenicol | L-arabinose | 0.5 mg/mL |
| pG-Tf2 | GroEL/ES, Trigger Factor | Chloramphenicol | L-arabinose | 0.5 mg/mL |
| pG-KJE8 | DnaK/DnaJ/GrpE, GroEL/ES | Chloramphenicol | L-arabinose | 0.5 mg/mL |
| pTf16 | Trigger Factor | Chloramphenicol | Tetracycline | 10 ng/mL |
Experimental Protocols
Protocol 1: Initial Co-expression Test for Solubility Enhancement
- Co-transform E. coli expression strain (e.g., BL21(DE3)) with the target protein plasmid and a chaperone plasmid (e.g., pKJE7 or pGro7).
- Grow colonies in double antibiotic media overnight.
- Inoculate main culture with double antibiotics. Grow at 37°C to OD600 ~0.6.
- Add chaperone inducer (e.g., 0.5 mg/mL L-arabinose for pKJE7). Grow for 1 hour at 37°C.
- Add target protein inducer (e.g., 0.1-0.5 mM IPTG). Shift temperature to 25°C or 30°C. Induce for 4-6 hours.
- Harvest cells by centrifugation. Lysis via sonication in suitable buffer.
- Separate soluble/insoluble fractions by high-speed centrifugation (15,000 x g, 30 min, 4°C).
- Analyze both fractions by SDS-PAGE to assess solubility shift.
Protocol 2: Assessing Folding & Activity Post-Solubilization
- After confirming solubility (Protocol 1, step 8), purify the soluble protein using affinity chromatography (e.g., His-tag).
- Measure specific activity of the purified protein using a defined enzymatic or binding assay.
- Compare the specific activity to a positive control (e.g., natively folded protein from another source).
- For GroEL/ES-assisted folding in vitro: Dialyze denatured, purified protein into refolding buffer containing GroEL, GroES, and an ATP-regenerating system. Monitor activity recovery over time.
Diagrams
Title: Chaperone Selection Decision Tree
Title: Chaperone Coordination Pathway in E. coli
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Chaperone Co-expression Studies
| Item | Function in Experiment | Example/Details |
|---|---|---|
| Chaperone Plasmid Kits | Express specific chaperone teams in E. coli. | Takara Bio's "Chaperone Plasmid Set" (pGro7, pKJE7, pG-Tf2, pTf16). |
| ATP, Magnesium Salt (MgCl2) | Essential cofactors for DnaK & GroEL/ES activity. | Add to lysis buffers (e.g., 1-5 mM ATP, 5-10 mM MgCl2) for in vitro folding assays. |
| Arabinose (L-Arabinose) | Inducer for chaperone expression from pBAD promoter. | Use high-purity grade. Typical working concentration: 0.1 - 0.5 mg/mL. |
| Protease Inhibitor Cocktail | Prevents degradation of target and chaperone proteins during lysis. | Use EDTA-free versions if studying metalloenzymes or Mg2+-dependent chaperones. |
| Detergents & Urea | For solubilizing inclusion bodies as a control/starting point. | Compare chaperone-assisted soluble yield vs. refolding from denaturant (e.g., 8M Urea). |
| Anti-Chaperone Antibodies | Validate chaperone overexpression via Western Blot. | Commercial antibodies for GroEL, DnaK, and Trigger Factor. |
| ATP Regeneration System | Maintains ATP levels in in vitro folding assays. | Phosphocreatine and creatine kinase. |
| Size-Exclusion Chromatography (SEC) | Analyze native oligomeric state and folding homogeneity post-purification. | Superdex series columns; compare retention volumes with/without chaperone co-expression. |
Welcome to the Technical Support Center for cultivation parameter optimization in recombinant protein expression research. This resource is designed to assist researchers in troubleshooting common issues, with a specific focus on improving enzyme solubility and yield within the context of overcoming solubility and expression challenges.
Frequently Asked Questions (FAQs) & Troubleshooting Guides
Q1: My recombinant enzyme is consistently forming inclusion bodies at 37°C. What temperature should I test to improve solubility? A: High expression rates at 37°C often overwhelm folding machinery. Implement a temperature reduction protocol.
- Troubleshooting Steps:
- Test Lower Temperatures: Induce expression at lower temperatures. A standard screening range is between 18°C and 30°C.
- Monitor Growth: Allow cultures to equilibrate at the lower temperature for 30-60 minutes before induction.
- Extend Expression: Double or triple the post-induction duration (e.g., from 4 hours to 16-24 hours at 20°C) to facilitate proper folding.
- Protocol: Temperature Shift Induction
- Inoculate primary culture and grow overnight at 37°C.
- Dilute secondary culture and grow at 37°C to an OD600 of ~0.6.
- Split culture into separate flasks for each temperature condition (e.g., 37°C, 25°C, 18°C).
- Transfer flasks to pre-cooled shakers set at target temperatures. Incubate for 1 hour.
- Induce all cultures with identical IPTG concentrations.
- Continue incubation at their respective temperatures for 16-20 hours.
Q2: At what cell density (OD600) should I induce expression to balance yield and solubility? A: Induction during mid-log phase is critical. Induction at too high a density can stress cells and reduce solubility.
- Troubleshooting Steps:
- Standard Point: Induce at an OD600 of 0.5-0.6 for most E. coli strains in rich media.
- For Toxic Proteins: Induce at a lower OD600 (0.3-0.4) to minimize stress from leaky expression before induction.
- For High-Density Cultures: For auto-induction media or specific protocols, follow validated OD600 targets, often up to 2.0-3.0 before temperature shift.
- Data Summary: Induction OD600 vs. Outcome
| Induction OD600 | Expected Protein Yield | Expected Solubility | Best Use Case |
|---|---|---|---|
| 0.3 - 0.4 | Lower | Higher | Toxic proteins, very sensitive enzymes |
| 0.5 - 0.6 | Moderate-High | Moderate-High | Standard recombinant expression |
| >0.8 | High (in dense culture) | Potentially Lower | Robust proteins, auto-induction schemes |
Q3: How does media formulation influence enzyme solubility, and what additives should I consider? A: Media affects metabolic burden, growth rate, and folding environment. Optimization is key.
- Troubleshooting Steps:
- Switch Media Type: If using rich media (e.g., TB, 2xYT) leads to aggregation, try a defined minimal medium (e.g., M9). Slower growth can improve folding.
- Add Folding Enhancers: Supplement with additives known to aid solubility.
- Adjust Carbon Source: For E. coli, try glycerol instead of glucose as a slower metabolizing carbon source.
- Protocol: Media Additive Screen for Solubility
- Prepare a base culture in your standard medium.
- At the time of induction, supplement aliquots with different additives from the reagent toolkit below.
- Express protein as per your optimized temperature/induction protocol.
- Lyse cells and separate soluble/insoluble fractions via centrifugation.
- Analyze both fractions by SDS-PAGE to compare solubility.
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Primary Function in Optimization |
|---|---|
| IPTG (Isopropyl β-D-1-thiogalactopyranoside) | Chemical inducer for lac/T7-based expression systems; concentration and timing are critical variables. |
| Autoinduction Media | Contains metabolizable sugars (e.g., lactose) that automatically induce expression at high cell density, useful for screening. |
| L-Arginine & L-Glutamate | Additives (0.4-0.8 M) that can act as chemical chaperones, reducing aggregation in vivo and in lysates. |
| Glycerol | Carbon source that promotes slower, more controlled growth compared to glucose; can be tested at 2-4% (v/v). |
| Protease Inhibitor Cocktails | Essential in lysis buffers to prevent degradation of soluble, especially unstable, recombinant enzymes. |
| Terrific Broth (TB) & Defined Minimal Media (M9) | Representative rich and minimal media types for testing growth rate's impact on protein folding. |
| Nickel-NTA or Cobalt Resin | For His-tagged protein purification; testing binding from soluble lysate indicates folded state. |
| Solubilization Buffer (Urea/Guanidine-HCl) | For denaturing and recovering protein from inclusion bodies as a last-resort comparison. |
Experimental Workflow & Pathway Diagrams
Title: Cultivation Parameter Optimization Workflow
Title: Stress Pathway to Inclusion Bodies vs. Solubility
Diagnosing and Solving Common Enzyme Solubility and Yield Problems
Technical Support Center
Troubleshooting Guides & FAQs
Q1: My SDS-PAGE gel shows no band for my expressed enzyme. What are the primary causes? A: This typically indicates a failure in expression or solubility. First, verify the optical density (OD600) and induction protocol. A significant increase in OD post-induction suggests growth but not necessarily protein expression. Run a gel of the total cell lysate (pellet) and the soluble fraction separately. If the band is only in the pellet, the protein is expressed but insoluble (inclusion bodies). Common causes include: incorrect induction temperature (too high), overly rapid induction, codon bias, or missing chaperones. Troubleshoot by lowering the induction temperature to 18-25°C, testing different inducer concentrations (e.g., 0.1-1.0 mM IPTG), and using an appropriate expression strain (e.g., BL21(DE3) pLysS, Origami B for disulfide bonds).
Q2: I have a strong band on my SDS-PAGE, but my activity assay shows negligible enzymatic activity. Why? A: The presence of a band confirms expression but not proper folding. This is a classic symptom of misfolded or inactive protein. First, check your assay conditions (pH, temperature, cofactors, substrate concentration) against literature values. If conditions are correct, the protein may be misfolded. Implement a refolding screen if the protein is insoluble, or co-express with chaperones (GroEL/GroES, DnaK/DnaJ/GrpE). For soluble protein, consider purifying under native conditions instead of denaturing conditions, and always include protease inhibitors and stabilizing agents (e.g., glycerol, DTT, specific ions) in your lysis and purification buffers.
Q3: My enzyme appears degraded on the gel, showing multiple lower molecular weight bands. How do I prevent this? A: Proteolytic degradation is common, especially with non-tagged proteins or those with flexible linkers. Immediately implement the following: 1) Perform lysis and all purification steps at 4°C. 2) Use a comprehensive protease inhibitor cocktail (e.g., containing AEBSF, EDTA, bestatin, etc.). 3) Consider adding 1-10 mM EDTA to inhibit metalloproteases. 4) Use an affinity tag (His-tag, GST) for faster purification. 5) If possible, shorten purification time or use an expression strain deficient in proteases (e.g., E. coli BL21(DE3) ompT lon).
Q4: What are the critical controls for an enzymatic activity assay following purification? A: Always run these controls in parallel: 1) No-Enzyme Control: Reaction mix with buffer instead of enzyme. Corrects for any non-enzymatic substrate conversion. 2) No-Substrate Control: Enzyme in assay buffer without substrate. Corrects for background signal from the enzyme/preparation. 3) Heat-Inactivated Enzyme Control: Boiled enzyme with substrate. Confirms activity is due to the protein. 4) Positive Control (if available): A known sample of active enzyme (commercial or previously validated). 5) Blank: All buffer components only, for spectrophotometer/plate reader zeroing.
Q5: How do I choose between a continuous and a discontinuous activity assay? A: The choice depends on your detection method and the enzyme's properties.
- Continuous Assay: Use if the reaction produces a detectable change (absorbance, fluorescence) in real-time. Preferred for kinetic studies (e.g., monitoring NADH oxidation at 340 nm).
- Discontinuous Assay (Endpoint): Use if no continuous signal is available or if you must quench the reaction to measure product (e.g., HPLC, mass spec). Requires multiple time points to establish linearity.
Research Reagent Solutions Toolkit
| Item | Function in Workflow |
|---|---|
| Laemmli Sample Buffer (2X) | Denatures protein samples for SDS-PAGE, includes tracking dye. |
| Precision Plus Protein Standards | Provides accurate molecular weight markers for SDS-PAGE analysis. |
| Polyacrylamide Gel (4-20% Gradient) | Separates proteins by size; gradient improves resolution of mixtures. |
| Protease Inhibitor Cocktail (EDTA-free) | Prevents proteolytic degradation during cell lysis and purification. |
| Ni-NTA Agarose Resin | Immobilized metal affinity chromatography resin for purifying His-tagged proteins. |
| PD-10 Desalting Columns | Rapid buffer exchange into assay-compatible buffer post-purification. |
| Bradford or BCA Assay Kit | Quantifies total protein concentration for normalizing activity assays. |
| 96-Well Clear Flat-Bottom Plates | Microplate for high-throughput spectrophotometric activity assays. |
| Cofactor (e.g., NADH, MgCl₂) | Essential component for many enzyme reactions; must be optimized. |
| Recombinant Lysozyme | Enhances bacterial cell wall lysis when added to lysis buffer. |
Table 1: Common Troubleshooting Parameters for Soluble Expression
| Parameter | Typical Test Range | Optimal Value (Varies) | Impact |
|---|---|---|---|
| Induction Temperature | 16°C, 25°C, 30°C, 37°C | Often 18-25°C | Lower temp slows growth, favors folding. |
| IPTG Concentration | 0.01 mM, 0.1 mM, 0.5 mM, 1.0 mM | Often 0.1-0.5 mM | Lower concentration can reduce burden. |
| Induction OD600 | 0.4-0.6, 0.8-1.0, >1.2 | 0.6-0.8 (mid-log) | Ensures healthy metabolic state. |
| Post-Induction Time | 3-4 h, 6 h, 16-20 h (o/n) | 4-6h at 37°C; O/N at 18°C | Balance between yield and degradation. |
Table 2: Key Components for a Standard Activity Assay Master Mix
| Component | Example Concentration | Purpose | Notes |
|---|---|---|---|
| Assay Buffer (Tris/HCl) | 50 mM, pH 7.5 | Maintains optimal pH | pH must be enzyme-specific. |
| Substrate | 1-10 x Km | Enzyme saturation | Determine Km first if unknown. |
| Cofactor | As required (e.g., 5 mM Mg²⁺) | Activates enzyme | Essential for many enzymes. |
| Enzyme Dilution | 1-100 µg/mL final | Provides linear reaction rate | Must be in linear range of assay. |
| Detection Reagent | Varies (e.g., 0.2 mM NAD⁺) | Generates detectable signal | Coupled assays common. |
Detailed Experimental Protocols
Protocol 1: Small-Scale Expression & Solubility Test (Trial Induction)
- Inoculation: Pick a single colony into 5 mL LB with antibiotic. Grow overnight at 37°C, 220 rpm.
- Dilution: Dilute the overnight culture 1:100 into 5 mL fresh LB + antibiotic in a 50 mL flask (total 5-10 mL per condition).
- Growth: Grow at 37°C, 220 rpm until OD600 ~0.6.
- Induction: Take a 1 mL pre-induction sample (pellet cells). Add IPTG to desired concentration. Split culture into flasks for different temperatures (e.g., 18°C, 25°C, 37°C).
- Harvest: Induce for 4-6 hours (or overnight for low temp). Take 1 mL post-induction sample. Pellet cells at 13,000 rpm for 1 min.
- Lysis & Fractionation: Resuspend pellet in 200 µL lysis buffer (with lysozyme & protease inhibitors). Freeze-thaw once or sonicate briefly. Centrifuge at 13,000 rpm for 15 min at 4°C. Carefully remove supernatant (soluble fraction). Resuspend pellet in 200 µL SDS-PAGE buffer (insoluble fraction).
- Analysis: Boil all samples (pre-induction, soluble, insoluble) for 5 min. Load 10-20 µL on an SDS-PAGE gel.
Protocol 2: Continuous Spectrophotometric Activity Assay (Example: Dehydrogenase)
- Prepare Enzyme: Dilute purified enzyme in assay buffer (containing stabilizing agents) to a working concentration. Keep on ice.
- Prepare Master Mix: For a 1 mL assay in a cuvette: 970 µL Assay Buffer (50 mM Tris-HCl, pH 8.0), 10 µL Substrate Stock (e.g., 100 mM final), 10 µL Cofactor Stock (e.g., 10 mM NAD⁺ final). Warm to assay temperature (e.g., 25°C) in spectrophotometer.
- Baseline: Pipette master mix into cuvette. Place in spectrophotometer, equilibrate for 1 min. Record absorbance at 340 nm for 60 sec to establish a stable baseline.
- Initiate Reaction: Add 10 µL of diluted enzyme to the cuvette. Mix quickly by inversion or pipetting.
- Data Acquisition: Immediately record the change in absorbance at 340 nm (A340) for 3-5 minutes. The slope of the linear portion (ΔA340/min) is used to calculate activity using NADH's extinction coefficient (ε340 = 6220 M⁻¹cm⁻¹).
Workflow and Pathway Diagrams
Diagram 1: Diagnostic Workflow for Enzyme Solubility & Activity
Diagram 2: Root Cause Analysis for Failed Enzyme Expression
Troubleshooting Guides & FAQs
Q1: After dilution refolding, my protein is aggregating and precipitating. What are the key parameters to adjust? A: Aggregation during dilution refolding is often due to a too-high protein concentration or an incorrect refolding buffer composition. Key adjustments include:
- Reduce Protein Concentration: Start refolding at 5-20 µg/mL. If successful, gradually increase in subsequent experiments.
- Optimize Redox System: For disulfide-bonded proteins, ensure an appropriate ratio of reduced (GSH) to oxidized (GSSG) glutathione (e.g., 1-10 mM GSH / 0.1-1 mM GSSG). A 10:1 to 5:1 ratio is common.
- Incorporate Additives: Include low concentrations of arginine (0.5-1 M) or glycerol (10-20% v/v) to suppress aggregation.
- Control pH and Temperature: Refold at 4-10°C and at a pH near the protein's pI, where solubility is often lowest but aggregation can be better managed.
Q2: During dialysis refolding, no protein is recovered in the final buffer. What could be the cause? A: Complete loss suggests precipitation on the dialysis membrane or within the tubing.
- Increase Denaturant Gradient: Do not dialyze directly from 8M urea/guanidine to zero. Use a stepwise dialysis protocol, decreasing denaturant concentration in steps of 1-2 M.
- Change Membrane Pore Size: Ensure the membrane's molecular weight cutoff (MWCO) is at least 2-3 times smaller than your protein's MW to prevent loss.
- Add Stabilizers: Include 0.5 M L-arginine and 5% glycerol in all dialysis steps to improve solubility during the transition.
- Monitor Precipitate: Perform dialysis with constant, gentle stirring. If precipitate forms in the dialysis bag, revert to a higher denaturant concentration and slow the gradient.
Q3: My protein elutes in the flow-through during on-column refolding using SEC. Why does this happen? A: Elution in the void volume indicates the protein is forming large aggregates that cannot enter the column resin pores.
- Pre-Filter Sample: Centrifuge and filter (0.22 µm) the solubilized inclusion body sample before loading to remove any pre-existing aggregates.
- Reduce Load Volume & Concentration: The load volume should be ≤ 2% of the column volume, and protein concentration should be < 1 mg/mL for SEC refolding.
- Optimize Loading Buffer: Ensure the loading buffer contains a denaturant concentration (e.g., 2-4 M urea) just sufficient to keep the protein monomeric but compatible with the SEC running buffer. A rapid, complete mixing at the column head is critical.
- Consider Alternative Method: On-column refolding by SEC is highly sensitive. If issues persist, perform batch refolding by dilution or dialysis first, then use SEC for purification/polishing.
Q4: How do I choose between redox systems (GSH/GSSG vs. cysteine/cystine vs. DTT redox pair) for disulfide bond formation? A: The choice depends on the protein's number of cysteines and sensitivity.
| Redox System | Typical Concentration Range | Best For | Considerations |
|---|---|---|---|
| GSH / GSSG | 1-10 mM GSH / 0.1-1 mM GSSG | Standard, robust refolding of proteins with 1-3 disulfide bonds. | Maintains a reducing potential; the 10:1 to 5:1 (GSH:GSSG) ratio is common. |
| Cysteine / Cystine | 2-5 mM Cysteine / 0.2-0.5 mM Cystine | Proteins requiring a stronger driving force for oxidation. | More potent but can lead to scrambling if not carefully controlled. |
| DTT Redox Pair | 1-5 mM DTTred / 0.1-0.5 mM DTTox | Research applications where a well-defined redox potential is needed. | Less common; requires preparation of oxidized DTT. |
General Tip: Start with GSH/GSSG. If refolding yield is low, test cysteine/cystine. Always use fresh, pH-adjusted stocks.
Table 1: Comparison of Core Refolding Method Parameters & Outcomes
| Method | Typical Protein Concentration | Total Volume | Time Required | Typical Yield Range | Key Advantage | Key Limitation |
|---|---|---|---|---|---|---|
| Dilution | 5-100 µg/mL | 0.1 - 10 L | 1-2 days | 20-60% | Simplicity, easy to scale up. | Large volumes, requires subsequent concentration. |
| Dialysis | 0.1-1 mg/mL | 0.01 - 0.5 L | 2-4 days | 10-50% | Gentle denaturant removal, no dilution. | Slow, prone to precipitation at interfaces. |
| SEC Chromatography | 0.1-1 mg/mL (load) | Column Dependent | 2-4 hours | 30-70% | Simultaneous refolding & purification, fast. | Complex setup, low total protein load. |
| HIC Chromatography | 0.5-2 mg/mL (load) | Column Dependent | 3-6 hours | 40-80% | Excellent for hydrophobic proteins, removes aggregates. | Requires optimization of salt gradient. |
Table 2: Common Refolding Buffer Additives & Their Roles
| Additive | Typical Concentration | Primary Function | Mechanism |
|---|---|---|---|
| L-Arginine HCl | 0.5 - 1.0 M | Suppress aggregation | Weakly interacts with unfolded polypeptide chains, suppressing non-productive aggregation. |
| Glycerol | 10 - 20% (v/v) | Stabilizer / Aggregation suppressor | Increases solution viscosity and stabilizes native protein structure. |
| GSH / GSSG | 1-10 mM / 0.1-1 mM | Redox system | Catalyzes formation and reshuffling of disulfide bonds to native state. |
| CHAPS / Triton X-100 | 0.1 - 2% (w/v) | Mild detergent | Solubilizes hydrophobic patches, preventing aggregation. |
| Sucrose / PEG | 0.2 - 0.5 M / 5-10% | Molecular crowder / stabilizer | Can enhance correct folding by excluded volume effect. |
Experimental Protocols
Protocol 1: Standard Dilution Refolding for a Disulfide-Bonded Protein Objective: To refold a protein from urea-solubilized inclusion bodies.
- Solubilization: Dissolve purified inclusion body pellet in Solubilization Buffer (8 M Urea, 50 mM Tris-HCl pH 8.0, 10 mM DTT) to a final concentration of 5-10 mg/mL. Incubate at room temperature for 1-2 hours with gentle agitation.
- Clarification: Centrifuge at 20,000 x g for 30 min at 15°C. Filter supernatant through a 0.22 µm filter.
- Refolding by Rapid Dilution: Prepare Refolding Buffer (50 mM Tris-HCl pH 8.0, 0.5 M L-Arginine, 1 mM GSH, 0.2 mM GSSG, 5% Glycerol). Chill to 4-10°C. With rapid stirring, add the solubilized protein dropwise to achieve a final protein concentration of 20-50 µg/mL.
- Incubation: Stir gently for 12-24 hours at 4-10°C.
- Concentration & Buffer Exchange: Concentrate the refolding mixture using a tangential flow filtration (TFF) system or centrifugal concentrator. Exchange into a suitable storage or assay buffer.
Protocol 2: Stepwise Dialysis Refolding Objective: Gentle denaturant removal for aggregation-prone proteins.
- Solubilization & Clarification: As per Protocol 1, steps 1-2.
- Primary Dialysis: Load the clarified solution into dialysis tubing (MWCO << protein MW). Dialyze against 100x volume of Dialysis Buffer 1 (4 M Urea, 50 mM Tris-HCl pH 8.0, 0.5 M L-Arginine, 1 mM GSH, 0.1 mM GSSG) for 6-12 hours at 4°C.
- Secondary Dialysis: Transfer the bag to Dialysis Buffer 2 (2 M Urea, other components identical to Buffer 1) for 6-12 hours at 4°C.
- Tertiary Dialysis: Transfer to Dialysis Buffer 3 (0 M Urea, 50 mM Tris-HCl pH 8.0, 0.2 M L-Arginine, 5% Glycerol) for 12-24 hours at 4°C. Change buffer once.
- Recovery: Retrieve the dialysate. Centrifuge at 10,000 x g for 20 min to remove any precipitate. Filter the supernatant (0.22 µm).
Protocol 3: On-Column Refolding & Purification via Size Exclusion Chromatography (SEC) Objective: Simultaneous refolding and initial purification.
- Column Equilibration: Equilibrate a HiLoad 16/600 Superdex 75 pg or similar SEC column with 2-3 column volumes (CV) of SEC Running Buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 M Urea, 5% Glycerol) at a flow rate of 0.5-1.0 mL/min.
- Sample Preparation: Solubilize and clarify protein as in Protocol 1. Then, dilute the sample with Solubilization Buffer (without DTT) to a final urea concentration matching the SEC Running Buffer (e.g., ~2 M) and a final protein concentration of ≤ 1 mg/mL. Filter (0.22 µm).
- Loading & Elution: Load a volume ≤ 2% of the column CV. Run isocratically with SEC Running Buffer. The denaturant gradient is formed in situ as the protein migrates through the column.
- Collection & Analysis: Collect the peak corresponding to the monomeric protein. Analyze by SDS-PAGE (non-reducing and reducing) and activity assays.
Diagrams
Diagram 1: Dilution Refolding Workflow
Diagram 2: Refolding Method Selection Logic
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Refolding |
|---|---|
| Urea & Guanidine HCl | Chaotropic agents used to denature and solubilize inclusion bodies by disrupting non-covalent interactions. |
| DTT (Dithiothreitol) | Strong reducing agent used during solubilization to break all incorrect disulfide bonds. |
| L-Arginine Hydrochloride | A chemical chaperone that suppresses aggregation of folding intermediates without stabilizing misfolded states. |
| Reduced/Oxidized Glutathione (GSH/GSSG) | A redox couple that creates a buffer system to catalyze the formation of correct disulfide bonds. |
| CHAPS Detergent | A zwitterionic detergent used to solubilize hydrophobic patches on folding intermediates, preventing aggregation. |
| Size Exclusion Chromatography Resin (e.g., Superdex) | For on-column refolding; separates monomeric protein from aggregates based on hydrodynamic radius. |
| Tangential Flow Filtration (TFF) System | For rapidly concentrating large, dilute volumes from dilution refolding into a usable protein solution. |
| Dialysis Tubing (Various MWCO) | For semi-permeable membrane-based slow denaturant removal during dialysis refolding. |
This technical support center addresses common experimental challenges in protein engineering efforts focused on modifying surface charge and hydrophobicity to improve enzyme solubility and expression, a core objective of broader thesis research in biocatalyst development.
FAQs & Troubleshooting Guides
Q1: After performing site-directed mutagenesis to introduce charged residues (e.g., Glu, Lys), my protein expression yield in E. coli drops drastically. What could be the cause? A: This is a common issue. A sudden drop in yield often indicates induced aggregation or toxicity.
- Troubleshooting Steps:
- Check Protein Solubility: Analyze the cell lysate supernatant (soluble) vs. pellet (insoluble) via SDS-PAGE. If your protein is in the pellet, the charge mutation may have caused non-native self-association.
- Reduce Expression Temperature: Lower the induction temperature (e.g., to 18-25°C) to slow protein synthesis and favor proper folding.
- Use a Solubility Tag: Temporarily fuse your protein to a well-folding tag (e.g., MBP, SUMO) to test if the core protein is now aggregation-prone. Co-express with chaperone plasmids (e.g., pG-KJE8).
- Re-evaluate Mutation Site: The introduced charge may be disrupting a critical, subtle interaction. Consider reverting and choosing an alternative site via homology modeling.
Q2: My computational tool (e.g., FoldX, Rosetta) suggested a set of hydrophobic-to-polar mutations to improve solubility, but the purified protein is inactive. Why? A: Loss of activity suggests the mutation is disrupting the functional fold or active site.
- Troubleshooting Steps:
- Verify Structural Context: Map the mutation onto a known structure (experimental or homology model). Even surface-facing hydrophobic residues can be part of a "hydrophobic patch" critical for stabilizing loop structures or subunit interfaces.
- Test Stability: Perform a thermal shift assay. A significant drop in melting temperature (ΔTm > 5°C) confirms destabilization.
- Employ a More Conservative Swap: Instead of mutating a hydrophobic residue to a charged one (e.g., Leu to Glu), try a milder polar residue (e.g., Leu to Ser or Gln) to reduce hydrophobic burial penalty while minimizing electrostatic repulsion risks.
Q3: How do I choose between rational design (targeted mutations) and semi-rational design (saturation mutagenesis) for optimizing surface properties? A: The choice depends on prior structural knowledge and the nature of the problem.
- Decision Guide:
- Use Rational Design when you have a high-confidence 3D structure and a clear, localized target (e.g., neutralizing a strong negative patch predicted by pI calculation, or breaking a specific hydrophobic cluster).
- Use Semi-Rational Design (e.g., focused saturation mutagenesis at multiple candidate positions) when the structural determinants of aggregation are diffuse or unknown. This is an exploratory strategy to sample sequence space.
Q4: My engineered variant shows improved soluble expression, but it precipitates during buffer exchange or concentration. What should I do? A: This indicates colloidal instability in the new solution condition.
- Troubleshooting Steps:
- Optimize Buffer Screen: Test different pH values (to alter protonation states), ionic strengths (to shield charges), and additives.
- Add Stabilizing Agents: Include low concentrations of non-ionic detergents (e.g., 0.01% Tween-20), arginine (0.1-0.5 M), or glycerol (5-10%).
- Avoid the Isoelectric Point: Ensure your buffer pH is at least 1.0 unit away from the predicted new pI of your variant to avoid precipitation at the pI.
Key Experimental Protocols
Protocol 1: In Silico Identification of Surface Patches for Mutagenesis
- Obtain your protein's 3D structure (PDB file or homology model).
- Use computational tools like PDB2PQR or the Adaptive Poisson-Boltzmann Solver (APBS) to calculate electrostatic surface potential.
- Visualize in PyMOL or Chimera. Identify large, contiguous patches of positive (blue) or negative (red) potential.
- Use tools like CamSol or Aggrescan3D to map predicted aggregation-prone hydrophobic regions on the protein surface.
- Select solvent-exposed residues (relative solvent accessibility > 25%) within these patches for mutagenesis. Prioritize residues not involved in catalysis or known binding interfaces.
Protocol 2: High-Throughput Screening for Solubility Using a GFP Fusion Assay
- Clone your mutant library into a vector where your protein is fused to the C-terminus of GFP (e.g., pET-GFP vectors).
- Express clones in 96-well deep-well plates by auto-induction at 25°C for 20-24 hours.
- Pellet cells and resuspend in lysis buffer (e.g., with lysozyme). Lyse by freeze-thaw or shaking with glass beads.
- Clarify lysates by centrifugation directly in the plate.
- Measure Total Fluorescence (ex. 488nm/em. 510nm) of the whole lysate (represents expression) and Soluble Fluorescence of the supernatant.
- Calculate a Solubility Index: Soluble Fluorescence / Total Fluorescence. Variants with a high index (>0.8) and high total fluorescence are primary hits.
Table 1: Common Charge Mutations and Their Typical Impact on Protein Properties
| Mutation Type | Example | Expected ΔpI* | Potential Solubility Impact | Risk of Destabilization |
|---|---|---|---|---|
| Negative Charge Introduction | Lys→Glu (K→E) | Decrease ~1.0 | Increase in basic buffer, Decrease at acidic pH | Low (if surface-exposed) |
| Positive Charge Introduction | Glu→Lys (E→K) | Increase ~1.0 | Increase in acidic buffer, Decrease at basic pH | Low (if surface-exposed) |
| Charge Reversal | Glu→Lys (E→K) | Increase ~2.0 | Highly context-dependent; can cause aggregation | Moderate to High |
| Charge Neutralization | Lys→Ala (K→A) | Decrease ~0.5 | May reduce solubility if charge network is broken | Moderate |
| Hydrophobic to Polar | Leu→Ser (L→S) | Minimal | Often increases solubility; can create new H-bonds | Low to Moderate |
| Polar to Hydrophobic | Ser→Leu (S→L) | Minimal | Often decreases solubility; promotes aggregation | High |
*ΔpI is approximate. Use tools like ExPASy's ProtParam for accurate calculation.
Table 2: Comparison of Computational Tools for Solubility Engineering Design
| Tool Name | Type | Primary Function | Best For | URL/Source |
|---|---|---|---|---|
| FoldX | Force Field | Predicts ΔΔG of mutation, alanine scanning, stability. | Rational design, prioritizing mutations for stability. | foldxsuite.org |
| Rosetta | Suite | High-end structural modeling & design (ddG, fixbb). | Semi-rational design, building combinatorial libraries. | rosettacommons.org |
| CamSol | Empirical Method | Predicts intrinsic solubility/profile of protein sequences. | Identifying aggregation-prone regions for targeting. | campsol.biocomp.unibo.it |
| Aggrescan3D | 3D Structure-Based | Maps aggregation "hot spots" onto protein 3D structures. | Targeting hydrophobic patches on the surface. | bioinf.uab.es/aggrescan3d/ |
| Poisson-Boltzmann Solver | Electrostatics | Calculates electrostatic potentials & pKa values. | Designing surface charge distributions. | poissonboltzmann.org |
Visualizations
Diagram 1: Decision Workflow for Mutagenesis Strategy
Diagram 2: Solubility Screening & Validation Pipeline
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Surface Engineering Experiments
| Item | Function & Rationale |
|---|---|
| KLD Enzyme Mix (NEB) | Enables rapid, high-efficiency site-directed mutagenesis following PCR by simultaneous kinasing, ligation, and DpnI digestion. |
| NNS Oligonucleotide Mix | For saturation mutagenesis. The NNS codon (N=A/T/G/C, S=G/C) covers all 20 amino acids with only 32 codons, reducing library bias. |
| pET-GFP Vectors | Allows fusion of target protein to GFP. GFP fluorescence serves as a direct, high-throughput reporter of soluble protein yield. |
| ArcticExpress (DE3) Cells | E. coli strain co-expressing chaperonins from a cold-adapted bacterium. Ideal for expressing difficult-to-fold mutants at low temps (12°C). |
| Thermal Shift Dye (e.g., SYPRO Orange) | A fluorescent dye used in Differential Scanning Fluorimetry (DSF) to measure protein melting temperature (Tm), indicating mutant stability. |
| HiTrap SP/CM or Q/DEAE Columns | Cation or anion exchange chromatography columns. Essential for purifying charge-engineered variants and analyzing elution behavior (salt concentration) as a proxy for surface charge change. |
| Zetasizer Nano (Malvern) | Dynamic Light Scattering (DLS) instrument. Critical for assessing aggregation state and hydrodynamic radius of purified variants in solution. |
High-Throughput Screening Approaches for Mutant Libraries and Expression Conditions
Troubleshooting Guides and FAQs
Q1: My 96-well plate expression culture shows no growth after induction. What could be wrong? A1: This is commonly due to plasmid loss or toxicity. Ensure your selective antibiotic (e.g., Kanamycin, Ampicillin) is present in the expression culture. Verify the inducer concentration (e.g., IPTG) is not toxic; perform a dose-response test from 0.01 mM to 1.0 mM. Check the integrity of your glycerol stock by streaking on a selective plate.
Q2: I observe high fluorescence background in my solubility screen using a GFP-fusion reporter, overwhelming the signal from soluble protein. A2: High background often stems from auto-fluorescence of media components or cell lysis. Switch to a defined, low-fluorescence medium like M9 minimal medium. Include a control well with cells expressing unfused GFP to establish a baseline. Ensure your centrifugation step post-lysis is sufficient (e.g., 4000 x g for 20 min) to pellet insoluble debris. Consider using a protease-deficient strain (e.g., BL21(DE3) lon/ompT) to reduce degradation-related artifacts.
Q3: My Bradford or colorimetric solubility assay results are inconsistent across plates. A3: Inconsistency is frequently an artifact of uneven cell lysis or temperature variation. Standardize lysis by using a multi-channel sonicator with a 96-well horn or a chemical lysis buffer with consistent lysozyme and detergent concentrations (e.g., 1 mg/mL lysozyme, 1% Triton X-100). Perform all assay steps with plates on a chilled, flat block to maintain uniform temperature. Include a standard curve of known soluble protein (e.g., BSA) on every plate for normalization.
Q4: When screening expression conditions, I see poor correlation between high solubility and high total protein yield. How should I prioritize hits? A4: Prioritize conditions that optimize the Solubility Fraction, not just total yield. Calculate: (Soluble Protein Concentration / Total Protein Concentration) x 100%. Conditions yielding a high total but low fraction (<20%) often produce inclusion bodies. Prioritize hits with a solubility fraction >50%, even if total yield is moderate, as this indicates properly folded protein more amenable to purification.
Q5: My robotic liquid handler is dispensing inconsistent volumes during library replication, leading to variable culture densities. A5: First, perform a calibration check using a dye (e.g., tartrazine) and a plate reader to measure dispensed volume accuracy. Ensure tips are properly seated and consider using conductive or low-retention tips for viscous glycerol stocks. Implement a "mix before aspirate" step in the protocol. If using metal pins for replication, clean and dry pins thoroughly between different library plates to avoid cross-contamination and droplet formation.
Table 1: Common Issues in HTS for Solubility and Expression
| Symptom | Possible Cause | Troubleshooting Action |
|---|---|---|
| No growth in specific wells | Plasmid instability, toxic mutation | Sequence plasmid from stock, reduce inducer concentration |
| Uniformly low yield across plate | Failed induction, degraded IPTG | Prepare fresh IPTG stock, check inducer addition log, verify temperature shift |
| High well-to-well variability | Inconsistent inoculation, evaporation | Use glass covers for plates, calibrate liquid handler, start cultures from fresh overnight colonies |
| Soluble signal saturated | Protein too soluble for assay range, over-expression | Dilute lysate sample, reduce induction time or temperature |
Detailed Experimental Protocols
Protocol 1: High-Throughput Solubility Screening via GFP-Fusion
Objective: To rapidly assess the solubility of protein variants from a mutant library expressed as C-terminal fusions to GFP.
- Clone mutant library into expression vector with C-terminal GFP tag (e.g., pET-EGFP).
- Transform into expression host (e.g., E. coli BL21(DE3)) and plate on selective agar. Pick colonies into 300 μL of LB medium with antibiotic in a 96-deep-well plate.
- Grow overnight at 37°C, 80% humidity, with shaking at 900 rpm.
- Sub-culture 30 μL of overnight culture into 1 mL of auto-induction medium (e.g., Overnight Express) in a new 96-deep-well plate.
- Express for 24 hours at 20°C with shaking.
- Harvest cells by centrifugation at 4000 x g for 15 min. Discard supernatant.
- Lysе pellets by resuspension in 200 μL of B-PER II (Thermo Scientific) or equivalent lysis buffer with lysozyme and DNAse I. Incubate with shaking for 15 min.
- Clarify lysate by centrifugation at 4000 x g for 30 min at 4°C.
- Transfer 100 μL of supernatant (soluble fraction) to a black, clear-bottom 96-well assay plate.
- Measure GFP fluorescence (Ex: 488 nm, Em: 510 nm) and total protein (e.g., by Bradford assay at 595 nm) on a plate reader.
- Calculate specific fluorescence (Fluorescence / A595) as a proxy for soluble protein yield.
Protocol 2: Multi-Factorial Expression Condition Screening
Objective: To identify optimal expression parameters (temperature, inducer concentration, time) for a target protein.
- Design a matrix of conditions in a 24-well block format. Variables: Temperature (16°C, 25°C, 37°C), IPTG concentration (0.1 mM, 0.5 mM, 1.0 mM), and induction time (4h, overnight).
- Inoculate a single fresh colony into 5 mL LB medium. Grow overnight at 37°C.
- Dilute culture 1:100 into 10 mL of fresh LB in each well of a 24-deep-well block.
- Grow at 37°C to mid-log phase (OD600 ~0.6).
- Induce by adding IPTG to the designated final concentrations and transfer blocks to the respective temperature-controlled shakers.
- Harvest cells at each time point by centrifugation.
- Lysе pellets via sonication or chemical lysis.
- Analyze total expression and solubility by SDS-PAGE (load normalized by OD600) and a solubility assay (as in Protocol 1).
- Score gels and solubility data to identify the condition yielding the highest soluble fraction.
Diagrams
Diagram 1: HTS Workflow for Solubility Optimization
Diagram 2: Key Factors in Protein Solubility
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for HTS Solubility Screening
| Item | Function | Example Product/Supplier |
|---|---|---|
| Auto-Induction Media | Allows high-density growth with automatic induction; reduces handling. | Overnight Express Instant TB Medium (MilliporeSigma) |
| GFP-Fusion Vectors | Reporter for solubility; fluorescence correlates with soluble fusion protein. | pET-EGFP series vectors (Addgene) |
| Chemical Lysis Reagent | Efficient, high-throughput lysis in plate format. | B-PER II Bacterial Protein Extraction Reagent (Thermo Scientific) |
| Lytic Enzyme Cocktail | Enhances lysis efficiency; reduces viscosity. | Lysozyme (Sigma), Benzonase Nuclease (MilliporeSigma) |
| 96-Well Deep-Well Plates | For high-cell-density culture with adequate aeration. | 2.2 mL square-well plates (e.g., Axygen) |
| Robotic Liquid Handler | For precise, reproducible pipetting across hundreds of samples. | Beckman Coulter Biomek i7 |
| Microplate Centrifuge | For pelleting cells and clarifying lysates in plate format. | Eppendorf Centrifuge 5810 R with plate rotor |
| Multimode Plate Reader | Quantifies fluorescence, absorbance for solubility and yield. | Tecan Spark or BioTek Synergy H1 |
| Protease-Deficient E. coli Strains | Minimizes target protein degradation. | BL21(DE3) lon/ompT (e.g., LOBSTR from Kerafast) |
| Solubility Enhancing Tags | Improves folding and solubility of target protein. | MBP (Maltose Binding Protein), SUMO, NusA tags |
Assessing Success: Techniques to Validate Solubility, Activity, and Structural Integrity
This technical support center is designed to support researchers within a broader thesis on addressing enzyme solubility and expression issues. Accurate determination of aggregation state is critical for characterizing soluble expression products.
FAQs & Troubleshooting Guides
Q1: My AUC sedimentation velocity data shows a very noisy baseline. What could be the cause? A: This is often due to micro-absorption effects from buffer components or a mismatch between the sample and reference buffer. Ensure precise buffer dialysis and match the reference buffer exactly. For enzymes, the presence of small amounts of precipitants or glycerol can cause this. Check the absorbance wavelength; 280 nm can be noisy, consider 230 nm or 250 nm if your protein allows.
Q2: During SEC-MALS, I observe a negative peak or a dip in the RI signal. How do I troubleshoot this? A: A negative RI peak typically indicates a lower refractive index in your sample zone compared to the mobile phase. This is common when your sample buffer has a different composition than the meticulously filtered mobile phase. Ensure your enzyme sample is in the exact same buffer as the mobile phase used for the run (dialyze vs. buffer exchange). Also, avoid using high concentrations of reducing agents like DTT (use TCEP instead) and detergents not present in the mobile phase.
Q3: My calculated molar mass from SEC-MALS is significantly higher than expected. What are the primary suspects? A: This directly indicates non-specific aggregation or a misfolded species.
- Suspect 1: Column Interaction. Your enzyme may be interacting with the SEC column matrix. Troubleshoot by altering buffer pH (to change charge) or adding 100-150 mM NaCl to shield electrostatic interactions. Confirm by running a different column chemistry (e.g., switch from agarose to polyacrylate).
- Suspect 2: Concentration-Dependent Aggregation. Re-run at multiple, lower loading concentrations (e.g., 0.5, 1, 2 mg/mL). If the measured molar mass decreases with concentration, you have reversible association. AUC is better suited to characterize this equilibrium.
- Suspect 3: Incomplete Solubility. The sample may contain large, insoluble aggregates that are filtered out or stuck in the lines, skewing the analysis of the soluble fraction. Always centrifuge and filter (0.1 µm) samples immediately before injection.
Q4: For AUC analysis of a heterogeneous solubility screen, how do I handle large numbers of samples efficiently? A: Use a 8-hole or 4-hole centerpiece in your rotor. Plan a "buffer screen" experiment where you load different buffer conditions (e.g., varying salt, pH, additives) into separate channels against a common reference. Use short solution columns (e.g., 80 µL sample, 120 µL reference) to reduce run time to 4-5 hours per velocity run at 50,000 RPM. Always include a well-characterized standard protein in one channel for calibration and diagnostic purposes.
Q5: When should I choose AUC over SEC-MALS for my solubility study? A: Refer to the decision table below.
Table 1: Method Comparison for Aggregation Analysis
| Parameter | Analytical Ultracentrifugation (AUC) | Size-Exclusion Chromatography with MALS (SEC-MALS) |
|---|---|---|
| Key Principle | Measures sedimentation under ultra-high gravity; first-principles. | Size separation followed by inline multi-angle light scattering. |
| Sample State | Analyzed in true solution state, no matrix. | Interacts with column stationary phase. |
| Concentration Range | Broad (µg/mL to mg/mL). Ideal for studying association equilibria. | Typically higher loading required (0.1-5 mg/mL). |
| Buffer Flexibility | High. Can use almost any biologically relevant buffer/additive. | Limited. Buffer must be compatible with column and free of light-scattering particles. |
| Detection of Reversible Aggregates | Excellent. Directly measures associating systems in equilibrium. | Poor. Dilution on column disrupts labile equilibria. |
| Analysis Speed | Slow (hours per sample, plus setup/cleaning). | Faster (~30 min per run, high-throughput autosampler possible). |
| Absolute Mass Requirement | No standard required. Based on first principles. | No standard required. MALS provides absolute mass. |
| Common Artifact Sources | Meniscus distortion, temperature fluctuations, window imperfections. | Column interactions, shear forces, filter retention of large aggregates. |
Experimental Protocols
Protocol 1: Basic SEC-MALS Experiment for Soluble Enzyme Analysis
- Buffer Preparation: Prepare and thoroughly degas (>30 min) and filter (0.1 µm) your chosen chromatography buffer (e.g., 20 mM HEPES, 150 mM NaCl, pH 7.4).
- Sample Preparation: Dialyze your clarified enzyme lysate or purified protein into exactly the same buffer. Centrifuge at 15,000 x g for 10 minutes and filter through a 0.1 µm centrifugal filter.
- System Equilibration: Equilibrate a suitable SEC column (e.g., Superdex 200 Increase) with at least 2 column volumes (CV) of filtered buffer at a constant flow rate (e.g., 0.5 mL/min). Connect the MALS and RI detectors according to manufacturer instructions and allow laser and detector signals to stabilize.
- Run & Data Collection: Inject 50-100 µL of sample (1-2 mg/mL recommended). Collect data from UV (280 nm), RI, and MALS (all angles) detectors. Perform a "blank" buffer injection to confirm baseline stability.
- Data Analysis: Use the instrument software to normalize MALS detectors, align peaks from different detectors, and calculate the absolute molar mass across the eluting peak. The slope of the molar mass vs. elution volume plot indicates homogeneity.
Protocol 2: AUC Sedimentation Velocity Experiment for Aggregation
- Sample & Reference Prep: Dialyze your enzyme sample into the desired buffer. Use the final dialysis buffer as the reference solution. Measure the exact density and viscosity of the buffer using a densitometer and viscometer, or calculate from known composition.
- Loading the Cell: Assemble the AUC cell with a 2-sector charcoal-filled epon centerpiece. Precisely load 420 µL of reference buffer into the reference sector. Load 400 µL of your sample into the sample sector. Ensure no bubbles are trapped.
- Experiment Setup: Place the cell in a rotor (e.g., An-50 Ti). Set the run parameters in the control software: Temperature = 20°C, Speed = 50,000 RPM, Scan Type = Absorbance (280 or 230 nm), Interval = 300 seconds, Duration = 8 hours.
- Data Acquisition: Start the run. The centrifuge will reach speed and begin collecting radial absorbance scans over time.
- Data Analysis: Use software like SEDFIT or Ultrascan. Model the data using the continuous c(s) distribution model. Input the correct buffer density, viscosity, and protein partial specific volume (calculate from sequence). The resulting distribution plot will show peaks corresponding to monomeric, dimeric, and higher-order aggregated species based on their sedimentation coefficients.
Visualizations
Title: AUC vs SEC-MALS Method Workflow Comparison
Title: Decision Guide: AUC or SEC-MALS for Aggregation?
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Aggregation State Analysis
| Item | Function & Importance |
|---|---|
| Ultra-Pure, Filtered Buffers | Essential for both methods to prevent scattering artifacts (SEC-MALS) and optical aberrations (AUC). Use 0.1 µm filtration. |
| TCEP (Tris(2-carboxyethyl)phosphine) | Preferred reducing agent over DTT/β-ME for SEC-MALS as it does not cause RI baseline shifts. Maintains enzyme cysteines in reduced state. |
| Standard Proteins (e.g., BSA, Thyroglobulin) | For SEC column calibration (MW standards) and AUC diagnostic checks (sedimentation coefficient standards). |
| High-Quality SEC Columns (e.g., Superdex, TSKgel) | Provide reproducible size separation with minimal non-specific interaction for a wide range of enzymes. |
| Charcoal-Filled Epon Centerpieces (AUC) | Standard centerpieces for absorbance measurements. Must be meticulously cleaned and inspected for scratches. |
| Precision Density & Viscosity Meter | Critical for accurate AUC data analysis to calculate correct sedimentation coefficients and molar masses. |
| 0.1 µm Centrifugal Filters | For final sample clarification immediately before injection in SEC-MALS to remove large aggregates and dust. |
| Compatible Detergents/Additives (e.g., CHAPS, GDN) | For studying membrane-associated enzymes or improving solubility without interfering with detection systems. |
Technical Support Center: Troubleshooting Protein Folding Validation Assays
Frequently Asked Questions (FAQs)
Q1: My Circular Dichroism (CD) spectrum shows very low ellipticity (flat line). What could be the cause? A: This is typically a buffer issue. The chosen buffer must be transparent in the far-UV range (<250 nm). High chloride concentration (e.g., from PBS or NaCl) absorbs strongly. Solution: Switch to a low-UV-absorbing buffer such as 10 mM sodium phosphate (pH 7.5) or 10 mM Tris-HCl, ensuring minimal salt. Check protein concentration; a concentration of at least 0.2 mg/mL in a 1 mm pathlength cell is typically required for far-UV CD.
Q2: In my Thermal Shift Assay (TSA), I see no transition (melting curve, Tm) even at high temperatures. A: This indicates the protein may not be folded, is aggregated, or the dye isn't binding. Troubleshooting Steps:
- Verify Dye Function: Use a control protein (e.g., lysozyme) with known stability.
- Check Aggregation: Pre-filter or centrifuge the sample to remove aggregates that quench dye signal.
- Optimize Dye:Protein Ratio: For SYPRO Orange, a final dilution of 5-10X from the stock (5000X) is standard, but test a range from 1X to 20X.
- Increase Protein Concentration: Use 0.2-5 µM protein in a 20 µL reaction.
Q3: My intrinsic tryptophan fluorescence shows no signal or only noise. A: This suggests either no tryptophan residues are present, they are fully quenched, or instrument settings are wrong. Solution:
- Check your protein sequence for Trp (W) residues.
- Ensure the correct excitation wavelength (~295 nm to avoid tyrosine contribution) is set and emission is scanned from ~310-400 nm.
- Verify the photomultiplier tube (PMT) voltage is set high enough (e.g., 600-700 V).
- Confirm the protein is properly folded; denatured proteins can have redshifted emission but still show signal.
Q4: The CD spectrum suggests high α-helical content, but the TSA shows a very low Tm (<40°C). Are these results contradictory? A: No. This is a common and critical finding in solubility/thesis research. It indicates your enzyme is folded but unstable under the assay conditions. The α-helical signal confirms secondary structure formation, while the low Tm indicates weak tertiary packing or lack of hydrophobic core stabilization. This directly informs your thesis work: you have a soluble, folded protein that requires optimization (e.g., buffer additives, point mutations) to achieve functional stability.
Experimental Protocols
Protocol 1: Far-UV Circular Dichroism for Secondary Structure
- Buffer Exchange: Dialyze or desalt purified protein into 10 mM sodium phosphate buffer, pH 7.5.
- Concentration: Measure A280 to determine concentration. Dilute to 0.2-0.5 mg/mL.
- Setup: Use a quartz cuvette with a 1 mm path length. Set instrument temperature to 20°C.
- Scan Parameters: Wavelength: 260-190 nm. Step resolution: 1 nm. Bandwidth: 1 nm. Averaging time: 2 seconds per point.
- Subtraction: Subtract the buffer-only spectrum from the protein spectrum.
- Analysis: Express data as mean residue ellipticity (degrees·cm²·dmol⁻¹) and analyze using algorithms like SELCON3 or K2D.
Protocol 2: Intrinsic Tryptophan Fluorescence for Tertiary Structure
- Sample Prep: Prepare protein at 0.1-0.5 mg/mL in a compatible buffer (avoid imidazole, DTT at high concentrations).
- Cuvette: Use a quartz fluorescence cuvette (semi-micro, 500 µL minimum).
- Excitation: Set excitation wavelength to 295 nm (bandwidth 5 nm).
- Emission Scan: Record emission spectrum from 310 to 400 nm (bandwidth 5 nm, scan speed medium).
- Denatured Control: Add a sample with 6 M GuHCl and re-scan. A redshift of ~20 nm indicates unfolding.
Protocol 3: Thermal Shift Assay (Differential Scanning Fluorimetry)
- Plate Setup: In a 96-well PCR plate, mix:
- 10 µL protein (final conc. 0.5-5 µM)
- 9 µL assay buffer
- 1 µL SYPRO Orange dye stock (5000X, dilute to 50X in buffer first, then add for a final 5X).
- Seal: Apply optical clear seal.
- Run: Place in real-time PCR instrument.
- Thermal Ramp: From 25°C to 95°C, increase at 1°C/min, with fluorescence measurement (ROX/FAM filter) at each step.
- Analysis: Plot fluorescence vs. temperature. Fit a Boltzmann sigmoidal curve to determine the inflection point (Tm).
Data Presentation
Table 1: Expected Spectral Signatures for Correctly Folded vs. Misfolded Protein
| Assay | Correctly Folded Signature | Misfolded/Aggregated Signature | Diagnostic Use |
|---|---|---|---|
| Far-UV CD | Double minima at ~208 nm & ~222 nm (α-helix). Minimum at ~217 nm (β-sheet). | Loss of minima, shifted peaks, or strong negative peak <200 nm (random coil). | Secondary structure content & type. |
| Intrinsic Fluorescence | Sharp peak with λmax ~330-340 nm (buried Trp). | Red-shifted λmax >350 nm (solvent-exposed Trp). Broadened peak. | Tertiary packing around aromatic residues. |
| Thermal Shift (Tm) | Clear sigmoidal transition. Tm >45°C often desired. | No transition, very low Tm (<40°C), or multiple transitions. | Thermal stability of tertiary structure. |
Table 2: Common Troubleshooting Metrics and Targets
| Problem | Parameter to Check | Target Value/Range |
|---|---|---|
| Low CD Signal | Protein Concentration | >0.2 mg/mL (1mm cell) |
| Pathlength | 0.1 mm or 1 mm for far-UV | |
| Noisy TSA | Dye Concentration | Final 2X - 10X from commercial stock |
| Protein Amount per Well | >0.5 µg | |
| Fluorescence Quenching | Scatter (Aggregation) | OD at 350 nm < 0.05 |
| Buffer Components | Avoid >1 mM DTT, imidazole |
Visualizations
Title: Protein Folding Validation Decision Workflow
Title: Linking Folding Assays to Solubility Research Thesis
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Folding Validation | Key Consideration for Solubility Research |
|---|---|---|
| SYPRO Orange Dye | Binds hydrophobic patches exposed upon protein denaturation in TSA. | Detects weak stability from poor folding; ideal for screening stabilizing buffers. |
| Low-UV CD Buffers (e.g., NaF, Na Phosphate) | Minimal absorbance in far-UV allows accurate secondary structure measurement. | Essential for characterizing soluble but marginally stable proteins. |
| Chaotropes (GuHCl, Urea) | Positive controls for denaturation in fluorescence and CD. | Define unfolded baseline; used to calculate free energy of folding (ΔG). |
| Fluorescence Cuvettes (Quartz, semi-micro) | Allow measurement of intrinsic tryptophan signal with small sample volumes. | Conserve precious soluble protein sample during optimization. |
| PCR Plates & Seals (Optically clear) | Used for high-throughput Thermal Shift Assays in real-time PCR machines. | Enables rapid screening of >96 buffer/additive conditions for stability. |
| Stabilizing Additives (e.g., Glycerol, Betaine, Ligands) | Can be added to assays to probe for increased Tm or corrected spectra. | Directly identifies formulation conditions that improve enzyme solubility & shelf-life. |
Troubleshooting Guides & FAQs
FAQ 1: My Recombinant Enzyme Shows No Detectable Activity. What Are the First Steps?
- Answer: Begin by systematically checking the expression and purification process.
- Confirm Expression: Run an SDS-PAGE gel of whole cell lysates (induced vs. uninduced) to verify protein production. Look for a band at the expected molecular weight.
- Check Solubility: Fractionate the lysate into soluble and insoluble (pellet) fractions. If your enzyme is in the pellet, it has formed inclusion bodies. See FAQ 2.
- Verify Purification: Analyze elution fractions from your affinity column (e.g., His-tag) by SDS-PAGE. Ensure the protein is present and relatively pure.
- Assay Buffer Compatibility: Ensure your activity assay buffer does not inhibit the enzyme (e.g., wrong pH, missing essential cofactors or divalent cations like Mg²⁺).
FAQ 2: My Enzyme is Insoluble (In Inclusion Bodies). How Can I Proceed with Kinetic Validation?
- Answer: You have two primary paths, refolding or solubility tagging.
- Path A: Refolding:
- Protocol: Solubilize inclusion bodies in 6-8 M Guanidine HCl or 8 M Urea. Use rapid dilution or slow dialysis into a refolding buffer (e.g., Tris-HCl, pH 8.0, low salt, redox couple like glutathione, arginine to prevent aggregation). Screen multiple refolding conditions.
- Note: Refolding is empirical and often yields low amounts of active protein, which may be sufficient for initial kinetic characterization.
- Path B: Alternative Solubility Tags: Switch from a small tag (His) to a large solubilizing fusion partner like Maltose-Binding Protein (MBP), GST, or SUMO. These can improve solubility during expression and can often be cleaved off later.
- Critical Consideration: Any tag or fusion partner must be removed before final kinetic comparison to the native enzyme, as it can affect enzyme structure and kinetics.
- Path A: Refolding:
FAQ 3: I Have Activity, But My Measured Km/Vmax Values Are Significantly Different from the Native Enzyme. How Do I Troubleshoot This?
- Answer: Discrepancies indicate a structural or environmental difference.
- Check Purity: Contaminating proteases or phosphatases can interfere. Use a more stringent purification step (e.g., size-exclusion chromatography).
- Verify Protein Concentration: Accurately determine active enzyme concentration, not just total protein (e.g., via quantitative Western blot, active site titration). Incorrect [E] directly skews Vmax calculations.
- Assay Conditions: Meticulously replicate the exact buffer, ionic strength, pH, and temperature conditions used for the native enzyme. Even small deviations matter.
- Post-Translational Modifications (PTMs): The native enzyme may have essential PTMs (phosphorylation, glycosylation) missing in your recombinant system. Consider using a different expression host (e.g., insect/baculovirus, mammalian cells).
- Incorrect Folding: The protein may be partially misfolded. Use circular dichroism (CD) spectroscopy to compare secondary structure with the native enzyme.
FAQ 4: What Are the Best Practices for Designing the Kinetic Assay Itself?
- Answer:
- Initial Velocity: Ensure you are measuring initial linear rates (less than 10% substrate conversion).
- Substrate Range: Use substrate concentrations spanning 0.2-5 x Km. Include enough points below Km.
- Controls: Include a negative control (no enzyme) and, if possible, a positive control (commercial or native enzyme).
- Replicates: Perform all measurements in technical and biological triplicate.
- Data Fitting: Use non-linear regression (e.g., Michaelis-Menten fit) in software like GraphPad Prism. Do not rely on linearized plots (Lineweaver-Burk) for parameter estimation.
Data Presentation: Typical Troubleshooting Outcomes
Table 1: Impact of Common Issues on Measured Kinetic Parameters
| Issue | Expected Effect on Km | Expected Effect on Vmax / Specific Activity | Root Cause |
|---|---|---|---|
| Protein Misfolding | Increase | Severe Decrease | Incorrect active site geometry. |
| Residual Solubility Tag | May Increase/Decrease | May Decrease | Steric hindrance or alteration of enzyme dynamics. |
| Missing Essential Cofactor | Apparent Increase | Severe Decrease | Reduced catalytic efficiency and substrate binding. |
| Non-Optimal Assay pH | Increase | Decrease | Altered protonation state of active site residues. |
| Contaminating Inhibitor | Increase | Decrease | Competition with substrate or allosteric inhibition. |
| Incorrect Enzyme Concentration | No Effect | Proportional Error | Vmax is directly miscalculated; Km remains accurate. |
Experimental Protocols
Protocol 1: Basic Michaelis-Menten Kinetics for Recombinant Enzyme Validation
Objective: Determine Km and Vmax for recombinant enzyme and compare to published native values.
Materials:
- Purified recombinant enzyme.
- Native enzyme (positive control, if available).
- Substrate stock solutions at varying concentrations.
- Assay Buffer (optimized for the enzyme).
- Microplate reader or spectrophotometer.
- GraphPad Prism or similar software.
Method:
- Prepare Reaction Mixes: In a 96-well plate, add assay buffer and varying concentrations of substrate. The final volume before enzyme addition should be 90 µL.
- Initiate Reactions: Start each reaction by adding 10 µL of appropriately diluted enzyme solution. Mix immediately.
- Measure Initial Rate: Record the change in absorbance/fluorescence over time (e.g., 5-10 minutes) at the recommended wavelength.
- Data Analysis: Convert raw data to reaction velocity (µM/min or nmol/min). Plot velocity (v) vs. substrate concentration [S]. Fit the data to the Michaelis-Menten equation:
v = (Vmax * [S]) / (Km + [S])using non-linear regression.
Protocol 2: Solubility Screening via Fractionation
Objective: Determine if recombinant protein is soluble or in inclusion bodies.
Method:
- Lysate Preparation: Resuspend cell pellet from 1 mL culture in 500 µL Lysis Buffer (e.g., 50 mM Tris pH 8.0, 150 mM NaCl, 1 mg/mL lysozyme, protease inhibitors).
- Sonication: Lyse cells on ice using sonication (3 pulses of 10 seconds each).
- Clarification: Centrifuge the lysate at 12,000 x g for 20 min at 4°C.
- Fractionation: Carefully separate the supernatant (soluble fraction). Resuspend the pellet in 500 µL of the same buffer + 1% SDS (insoluble fraction).
- Analysis: Load equal volume equivalents of the total lysate, soluble fraction, and insoluble fraction on an SDS-PAGE gel to visualize distribution.
Mandatory Visualization
Title: Troubleshooting Workflow for Enzyme Kinetic Validation
Title: Kinetic Assay Data Generation and Analysis Steps
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Recombinant Enzyme Kinetic Studies
| Item | Function & Rationale |
|---|---|
| pET Vector Systems | Common E. coli expression vectors with strong T7 promoters for high-yield protein production. |
| Rosetta(DE3) Cells | E. coli strains supplying rare tRNAs for genes with mammalian codon bias, improving expression. |
| Maltose-Binding Protein (MBP) Tag | Large solubility-enhancing fusion partner; often increases chances of soluble expression. |
| HisTrap HP Column | Standard nickel-affinity chromatography column for rapid purification of His-tagged proteins. |
| TEV Protease | Highly specific protease for removing affinity tags, leaving a native or minimal residual sequence. |
| Size-Exclusion Chromatography (SEC) Standard | Protein standards (e.g., Biorad Gel Filtration Standard) to calibrate SEC columns for purity and oligomerization state analysis. |
| Continuous Kinetics Assay Kit (e.g., NADH-linked) | Coupled enzyme systems to conveniently monitor dehydrogenase/kinase activity by NADH absorbance. |
| Bradford/BCA Assay Reagents | For determining total protein concentration. Note: Requires a standard curve from the same protein if possible. |
| Active Site Titration Inhibitor | A tight-binding, irreversible inhibitor to determine the concentration of active enzyme, not just total protein. |
Technical Support Center: Troubleshooting Solubility & Expression
FAQs & Troubleshooting Guides
Q1: My recombinant protein is entirely in the inclusion body fraction after expression in E. coli. What are my primary strategies for recovery? A: You have three main strategic pathways: 1) Refolding in vitro: Solubilize the inclusion body with a strong denaturant (e.g., 8M urea or 6M guanidine-HCl), then refold by dialysis or dilution. Success rates vary from 10-50% and are highly protein-dependent. 2) Solubilization with mild detergents or high-pH buffers: Use sarkosyl or alkaline buffer (pH 10.5-12) to extract protein while retaining some structure. 3) Switch to a different expression system: Consider bacterial strains with chaperone co-expression (e.g., pGro7 plasmid), cell-free systems, or eukaryotic hosts like Pichia pastoris or insect cells.
Q2: During refolding, my protein precipitates upon removal of the denaturant. How can I optimize conditions? A: This is a common aggregation event. Systematically test the following:
- Reduced protein concentration during refolding (10-50 µg/mL).
- Different refolding buffers: Incorporate arginine (0.4-0.8 M), glycerol, or non-denaturing chaotropes to suppress aggregation.
- Stepwise dialysis versus rapid dilution to identify the optimal denaturant removal rate.
- Temperature: Perform refolding at 4°C to slow kinetics and reduce aggregation.
- Use of redox shuffling systems (GSH/GSSG) if disulfide bonds are present.
Q3: I am using detergent solubilization for a membrane protein, but it becomes inactive. What could be wrong? A: Detergent choice is critical for preserving function. The issue likely stems from:
- Destabilizing the native lipid environment: Try a milder detergent (e.g., DDM, LMNG) instead of harsh ionic ones (e.g., SDS, CTAB).
- Incorrect Critical Micelle Concentration (CMC): Maintain detergent concentration above its CMC during purification to prevent protein aggregation.
- Stripping essential cofactors: Ensure your buffer includes necessary lipids, ions, or stabilizing ligands throughout the process.
Q4: How effective is fusion tag-assisted solubility enhancement, and which tag should I choose? A: Fusion tags can improve solubility by 30-70%. Common choices and their trade-offs are summarized in Table 1. A key troubleshooting step is to test cleavage: if the protein precipitates after tag removal, consider using a milder cleavage condition or a different linker sequence.
Q5: My protein is soluble but forms high-order aggregates. How can I address this? A: Soluble aggregates often indicate partially misfolded species.
- Analyze by size-exclusion chromatography (SEC) coupled with multi-angle light scattering (MALS).
- Introduce stabilizing additives like trimethylamine N-oxide (TMAO) or specific salts.
- Screen for optimal pH and ionic strength to maximize monodispersity.
- Consider site-directed mutagenesis to surface-exposed hydrophobic patches predicted by in silico tools.
Table 1: Comparison of Solubilization Strategy Success Rates & Trade-offs
| Strategy | Typical Success Rate (Soluble, Active Protein) | Key Advantages | Major Trade-offs/Limitations |
|---|---|---|---|
| In Vitro Refolding | 15-40% | Applicable to large yields from IBs; purifies from host proteins. | Low throughput; condition-specific; often low final activity. |
| Fusion Tags (e.g., MBP, GST, SUMO) | 50-70% | High solubility enhancement; facilitates purification. | May not work for all proteins; tag can interfere with function/struct. |
| Chaperone Co-expression | 40-60% | In vivo folding aid; can improve activity. | Increased metabolic load; variable effect; requires optimization. |
| Detergent Solubilization | 30-50% (Membrane Proteins) | Essential for membrane proteins; can maintain native state. | Detergent removal challenging; can inactivate proteins; costly. |
| Expression Host Switching | 60-80% (for difficult proteins) | Access to PTMs; better folding machinery. | Significantly higher cost & time; lower expression yield possible. |
Table 2: Common Refolding Additives & Their Effects
| Additive | Typical Concentration | Primary Function | Considerations |
|---|---|---|---|
| L-arginine-HCl | 0.4 - 0.8 M | Suppresses aggregation; stabilizes intermediates. | Can interfere with ion-exchange chromatography. |
| Glycerol | 10-20% (v/v) | Stabilizes native state; reduces hydrophobic interactions. | High viscosity can complicate handling. |
| GSH/GSSG Ratio | 1-10 mM / 0.1-1 mM | Catalyzes disulfide bond reshuffling. | Must be optimized for each protein; pH-sensitive. |
| CHAPS | 1-10 mM | Mild detergent, prevents aggregation. | Can be difficult to remove. |
Detailed Experimental Protocols
Protocol 1: High-Throughput Screening for Refolding Buffers
- Solubilize inclusion bodies in 8M urea, 50 mM Tris-HCl, 10 mM DTT, pH 8.0, for 1 hour at room temperature.
- Clarify by centrifugation at 16,000 x g for 20 minutes.
- Dilute the denatured protein 50-fold into a 96-well plate containing different refolding buffers (varying pH, salts, additives like arginine, glycerol, redox pairs).
- Incubate at 4°C for 24-48 hours.
- Assay for solubility (via absorbance at 340 nm for turbidity) and activity (via specific enzymatic or binding assay).
- Scale-up the most promising condition for dialysis and further purification.
Protocol 2: Detergent Screening for Membrane Protein Solubilization
- Harvest and lyse cells expressing the membrane protein.
- Prepare membrane fraction via ultracentrifugation (100,000 x g, 1 hour).
- Aliquot membrane pellets into microcentrifuge tubes.
- Resuspend each pellet in different screening buffers containing 1% (w/v) of various detergents (e.g., DDM, OG, Triton X-100, LDAO, Fos-Choline-12). Include a buffer-only control.
- Incubate with gentle rotation for 2-3 hours at 4°C.
- Separate soluble and insoluble fractions via ultracentrifugation (100,000 x g, 30 min).
- Analyze supernatant (soluble) and pellet (insoluble) fractions by SDS-PAGE and Western blot to determine solubilization efficiency.
Visualizations
High-Throughput Refolding Screening Workflow
Decision Tree for Solubilization Strategy Selection
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function in Solubilization/Expression |
|---|---|
| pET Vector Series | High-copy number T7 expression vectors for strong, inducible protein production in E. coli. |
| Rosetta(DE3) Cells | E. coli strains supplying rare tRNAs for genes with codons poorly used in bacteria. |
| Chaperone Plasmids (pGro7, pKJE7) | Plasmids for co-expressing GroEL/ES or DnaK/DnaJ/GrpE chaperone systems to aid folding. |
| HisTrap HP Column | Immobilized metal affinity chromatography (IMAC) column for purifying His-tagged proteins. |
| Detergents: DDM, LMNG | Mild, non-ionic detergents for stabilizing membrane proteins during extraction and purification. |
| SUMO Protease | Highly specific protease for cleaving SUMO fusion tags, often yielding native N-termini. |
| Size-Exclusion Columns (Superdex) | For analyzing oligomeric state, removing aggregates, and buffer exchange into final formulation. |
| HaloTag | Fusion tag that covalently binds to solid supports, allowing stringent washing before tag cleavage. |
Conclusion
Successfully addressing enzyme solubility and expression requires a multi-faceted approach, integrating fundamental knowledge of protein biophysics with systematic methodological experimentation and rigorous validation. Key takeaways include the importance of early diagnostic workflows, the strategic combination of fusion tags and chaperone systems, and the necessity of functional assays beyond simple solubility measurements. Future directions point toward machine learning for solubility prediction, advanced cell-free expression systems, and engineered host strains tailored for difficult-to-express proteins. Mastering these challenges is pivotal for accelerating the development of enzyme-based therapeutics, diagnostics, and industrial biocatalysts, directly impacting progress in personalized medicine and green chemistry.