Unlocking Azetidine Biosynthesis: The Catalytic Power of Non-Haem Iron Enzymes in Drug Discovery
This article explores the recent groundbreaking discovery of a novel biosynthetic pathway for azetidine-containing amino acids, a class of compounds with significant pharmaceutical potential.
Unlocking Azetidine Biosynthesis: The Catalytic Power of Non-Haem Iron Enzymes in Drug Discovery
Abstract
This article explores the recent groundbreaking discovery of a novel biosynthetic pathway for azetidine-containing amino acids, a class of compounds with significant pharmaceutical potential. We delve into the mechanism by which non-haem iron-dependent enzymes, specifically PolF from the HDO superfamily and its helper PolE, catalyze the formation of the highly strained azetidine ring from linear amino acid precursors like L-isoleucine. Covering foundational concepts, methodological approaches, and troubleshooting strategies, this resource provides researchers and drug development professionals with a comprehensive understanding of this enzymatic process. The content also validates this pathway against known mechanisms and discusses its broader implications for the efficient synthesis of bioactive molecules and the development of new antifungal agents.
The Azetidine Enigma: From Bioactive Motif to Biosynthetic Breakthrough
Azetidines, four-membered saturated nitrogen-containing heterocycles, are increasingly significant scaffolds in medicinal chemistry and organic synthesis. Their importance stems from a combination of unique structural properties, substantial ring strain, and presence in bioactive molecules. This guide provides a technical overview of azetidine characteristics, explores a novel biosynthetic pathway relevant to azetidine amino acid production, and details contemporary synthetic methodologies.
Core Structural Properties and Significance in Drug Discovery
The azetidine ring is a cyclobutane derivative where one carbon atom is replaced by nitrogen. This substitution creates a polar, strained heterocycle with distinct physicochemical properties.
Ring Strain and Conformation: The bond angles within the azetidine ring are constrained to approximately 90°, a significant deviation from the ideal tetrahedral geometry of sp³-hybridized atoms (109.5°). This distortion results in a ring strain energy of about 105 kJ molâ»Â¹ [1]. This strain is a primary driver of azetidine reactivity, facilitating ring-opening and ring-expansion reactions. The ring adopts a "wing-shaped" conformation, and the introduction of substituents can significantly influence its overall geometry and steric profile [2].
Physicochemical Advantages: Incorporating azetidines into bioactive molecules is a established strategy for improving drug-like properties. Azetidines can increase molecular rigidity, which reduces the entropic penalty upon binding to a biological target. They often improve aqueous solubility and enhance metabolic stability by blocking sites of oxidative metabolism. Furthermore, as saturated rings, they help increase the fraction of Csp³ character (Fsp³) in a molecule, a parameter associated with higher clinical success rates in drug development [3].
Presence in Bioactive Molecules: Azetidine is a key pharmacophore in several approved drugs and clinical candidates. Notable examples include:
- Cobimetinib: A mitogen-activated protein kinase (MEK) inhibitor used in oncology [4].
- Azelnidipine: A dihydropyridine calcium channel blocker used as an antihypertensive agent [4] [2].
- Baricitinib: A Janus kinase (JAK) inhibitor used for autoimmune diseases [2].
- Ximelagatran: An oral direct thrombin inhibitor [4].
A Novel Biosynthetic Pathway for Azetidine Amino Acids
While traditional chemical synthesis of azetidines is challenging, nature has evolved enzymatic pathways to construct this strained ring. Recent groundbreaking research has elucidated a novel biosynthetic route to azetidine-containing amino acids in the polyoxin antifungal pathway, mediated by non-haem iron-dependent enzymes [5] [6] [7].
Experimental Protocol: Elucidating the PolF and PolE Pathway
The following methodology outlines the key experiments used to characterize this biosynthesis.
Gene Knockout and Metabolite Analysis: The genes encoding putative biosynthetic enzymes PolE and PolF were individually disrupted in the polyoxin producer Streptomyces cacaoi via in-frame deletion [5].
- Analysis: The resulting mutant strains were cultured under polyoxin-producing conditions. The fermentation broth was analyzed using liquid chromatography–mass spectrometry (LC-MS). The polF mutant did not produce any measurable polyoxin A (<1% of wild-type), demonstrating its essential role. The polE mutant produced a reduced amount (~10% of wild-type), indicating its auxiliary function [5].
Enzyme Expression and Purification: The polF gene was expressed in E. coli, and the resulting protein was purified. The enzyme was initially isolated in an apo (metal-free) form [5].
In Vitro Reconstitution of Activity:
- Metallation: Apo-PolF was incubated with an excess of Fe(II) under anaerobic conditions. Unbound iron was removed via a desalting column, resulting in Fe-reconstituted PolF [5].
- Assay Conditions: Enzyme assays were performed by incubating PolF with L-isoleucine (L-Ile) or L-valine (L-Val) under anaerobic conditions. Reactions were initiated by adding an Oâ‚‚-saturated buffer. The dependence on Oâ‚‚ and Fe(II) was confirmed through control experiments omitting each component [5].
- Product Analysis: Reaction products were derivatized with dansyl chloride (DnsCl) and analyzed by LC-MS. The azetidine product from L-Ile was identified as polyoximic acid (PA) by comparison with an authentic standard. The product from L-Val was identified as 3-methyl-ene-azetidine-2-carboxylic acid (MAA) through NMR characterization [5].
Mechanistic Probing via Intermediate Trapping: Assays with L-Val under single-turnover conditions (using 2 equivalents of Fe(II) and no external reductant) allowed for the detection and quantification of reaction intermediates, including 3,4-dehydrovaline (3,4-dh-Val) and 3-dimethylaziridine-2-carboxylic acid (Azi) [5].
Biosynthetic Mechanism and Pathway Logic
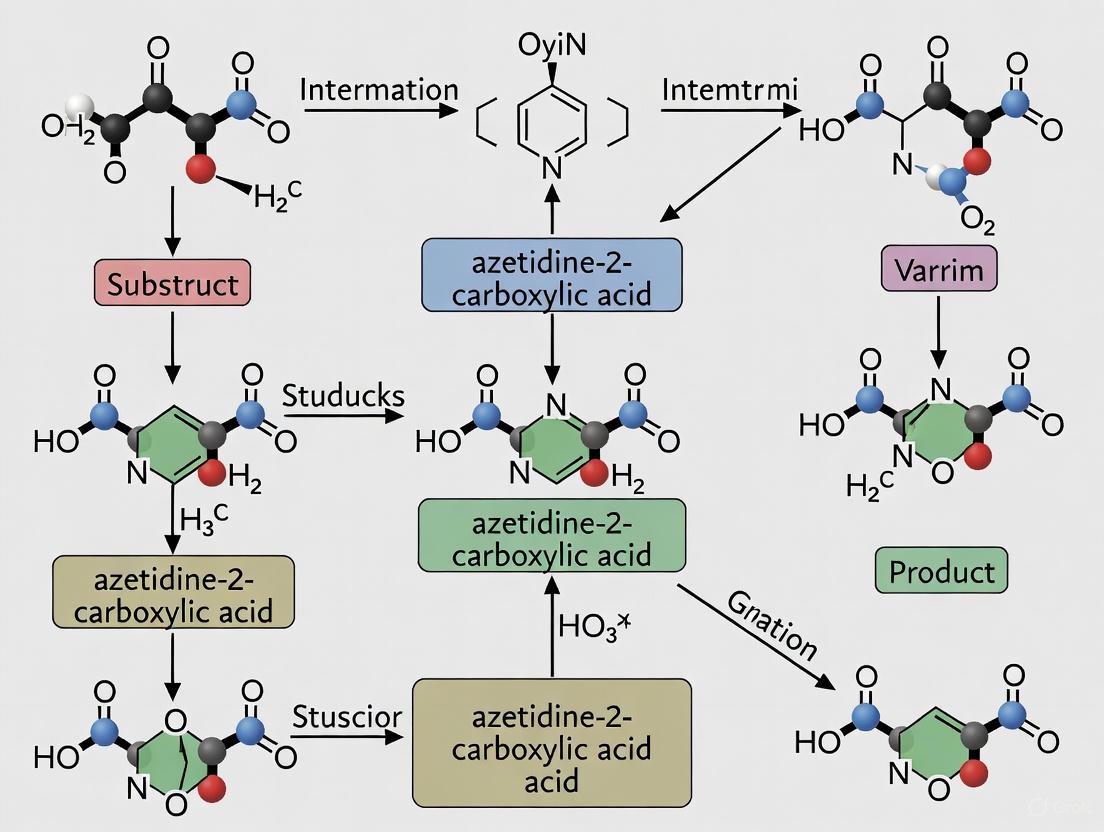
The biosynthesis of polyoximic acid from L-Ile is a two-step enzymatic process. The following diagram illustrates the roles of PolE and PolF in this pathway.
Role of PolE: PolE is an Fe and pterin-dependent oxidase that catalyzes the desaturation of L-Ile to form a 3,4-dehydroisoleucine intermediate. This step increases the flux through the pathway but is not strictly essential, as PolF can process the native amino acid directly at a lower efficiency [5] [7].
Role of PolF: PolF is the key enzyme responsible for azetidine ring formation. It belongs to the haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily. It utilizes a diiron (Feâ‚‚) core to activate molecular oxygen [5] [7].
- Mechanism: The current model suggests PolF generates a μ-peroxo-Fe(III)₂ intermediate capable of cleaving an unactivated C–H bond. The subsequent transformations, including the crucial intramolecular C–N bond formation to close the four-membered ring, are proposed to proceed through radical mechanisms [5]. The enzyme can catalyze multiple reactions on a single substrate—desaturation, hydroxylation, and C–N bond formation—with the azetidine-forming pathway being dominant for L-Ile and L-Val [5].
Quantitative Profile of PolF Substrate Specificity
The substrate specificity of PolF was experimentally determined, revealing its preference for medium-chain aliphatic amino acids. The table below summarizes the products formed from various substrates.
Table 1: Substrate Specificity and Product Profile of PolF Enzyme
| Substrate | Primary Product(s) | Product Type | Key Observation |
|---|---|---|---|
| L-Isoleucine | Polyoximic Acid | Azetidine | Native biosynthetic substrate [5] |
| L-Valine | 3-Methylene-azetidine-2-carboxylic Acid | Azetidine | Efficient conversion to azetidine [5] |
| L-Leucine | Hydroxylation (major), Desaturation (minor) | Non-cyclic | No azetidine formed [5] |
| L-Methionine | Hydroxylation (major), Desaturation (minor) | Non-cyclic | No azetidine formed [5] |
| L-allo-Ile, D-Ile, D-allo-Ile | Azetidine | Azetidine | Small but detectable amounts; stereochemistry not absolute [5] |
Modern Synthetic Approaches to Azetidines
Beyond biosynthesis, organic chemists have developed innovative synthetic strategies to access diverse azetidine scaffolds. Three contemporary methods are highlighted below.
Intermolecular Aza Paternò-Büchi Reaction
This photochemical [2+2] cycloaddition between imines and alkenes is a direct and modular approach. A recent breakthrough uses N-sulfamoyl fluoride-substituted imines, which, upon triplet energy transfer catalysis, generate reactive intermediates that couple with a wide range of alkenes to form azetidines in high yields [3]. The N-SFâ‚‚ group is a valuable handle for further functionalization or removal.
Strain-Release Difunctionalization of Azabicyclo[1.1.0]butanes (ABBs)
Highly strained ABBs serve as versatile precursors to 1,3-disubstituted azetidines. Under photoredox catalysis, these spring-loaded scaffolds undergo ring-opening difunctionalization. For example, using a bench-stable SFâ‚…-transfer reagent, ABBs can be converted to N-SFâ‚… azetidines, a novel class of potential bioisosteres with high aqueous stability and increased lipophilicity [8].
Enantioselective Difunctionalization of Azetines
A powerful catalytic method provides access to chiral 2,3-disubstituted azetidines. Using a Cu/bisphosphine catalyst system, azetines undergo asymmetric borylallylation, installing two versatile functional groups with concomitant creation of two stereogenic centers in a single step with excellent regio-, enantio-, and diastereocontrol [9]. The boryl and allyl groups can be further transformed, enabling rapid diversification.
The Scientist's Toolkit: Key Research Reagents and Materials
Table 2: Essential Reagents for Azetidine Biosynthesis and Synthesis Research
| Reagent / Material | Function in Research | Application Context |
|---|---|---|
| Fe(II) Salts (e.g., FeSOâ‚„) | Cofactor for non-haem iron enzymes (PolE, PolF) [5]. | In vitro enzymatic assays |
| Bâ‚‚pinâ‚‚ (Bis(pinacolato)diboron) | Boron source for enantioselective borylallylation reactions [9]. | Synthetic chemistry: Cu-catalyzed difunctionalization |
| ABB Precursors (Azabicyclo[1.1.0]butanes) | Spring-loaded intermediates for strain-release synthesis [8]. | Synthetic chemistry: Photocatalytic synthesis of N-SFâ‚… azetidines |
| SFâ‚… Transfer Reagents (Bench-stable) | Source of SFâ‚… radical for pentafluorosulfanylation [8]. | Synthetic chemistry: Radical difunctionalization of ABBs |
| Sulfamoyl Fluoride Imines | Substrates for generating reactive triplet imine intermediates [3]. | Synthetic chemistry: Aza Paternò-Büchi reaction |
| Chiral Bisphosphine Ligands (e.g., Ph-BPE) | Control enantioselectivity in metal-catalyzed reactions [9]. | Synthetic chemistry: Asymmetric synthesis of azetidines |
| Photocatalysts (e.g., 2,7-Brâ‚‚-TXT) | Mediate energy or electron transfer under light irradiation [3] [8]. | Synthetic chemistry: Photochemical cyclizations and radical reactions |
| Fmoc-D-Pro-OH | Fmoc-D-Pro-OH, CAS:101555-62-8, MF:C20H19NO4, MW:337.4 g/mol | Chemical Reagent |
| Fmoc-1-Nal-OH | Fmoc-1-Nal-OH, CAS:96402-49-2, MF:C28H23NO4, MW:437.5 g/mol | Chemical Reagent |
Azetidine, a saturated four-membered ring containing one nitrogen atom, is a structure of significant interest in medicinal chemistry due to its substantial ring strain (approximately 25.4 kcal molâ»Â¹) and presence in numerous bioactive compounds [5] [10]. This high-energy configuration imparts unique reactivity and conformational properties that make it valuable for drug design, particularly in the development of antimicrobial agents [7]. Despite its pharmaceutical importance, the biosynthetic origins of azetidine-containing natural products have remained largely enigmatic until recent investigations elucidated novel enzymatic pathways [5] [6].
The antifungal nucleoside polyoxin A contains a distinctive azetidine amino acid known as polyoximic acid (PA) [5] [10]. Early isotope-labeling studies indicated that PA originates from l-isoleucine (l-Ile), suggesting a previously uncharacterized biosynthetic mechanism distinct from known pathways [10]. Two previously unidentified enzymes, PolE and PolF, encoded within the polyoxin biosynthetic gene cluster, have now been identified as the key catalysts responsible for azetidine ring formation [5]. This review comprehensively examines the mechanistic details of this recently discovered biosynthetic pathway, with particular emphasis on the novel non-haem iron-dependent enzymes that construct this strained heterocycle.
Biosynthetic Pathways to Azetidine Rings
Previously Characterized Pathways
Before the elucidation of the polyoxin pathway, two primary enzymatic mechanisms were known to generate azetidine rings in natural products:
S-adenosyl-l-methionine (SAM)-dependent enzymes: These catalysts promote an intramolecular nucleophilic cyclization of SAM, yielding azetidine carboxylic acid and 5'-methylthioadenosine [5] [10]. This pathway represents a well-established route to azetidine-containing amino acids.
α-ketoglutarate (α-KG) and Fe-dependent oxygenases: Exemplified by the okaramine biosynthetic pathway, these enzymes facilitate radical-mediated oxidative C–C bond formation to generate the azetidine ring [5] [10]. This mechanism expands the repertoire of radical enzymes in natural product biosynthesis.
Both these established routes share a common limitation: their dependence on metabolically expensive precursors (SAM or α-KG), which restricts their utility for biocatalytic applications [5] [10].
The Polyoxin Azetidine Biosynthetic Pathway
The biosynthesis of polyoximic acid in the antifungal polyoxin pathway represents a distinct mechanistic paradigm. Gene knockout experiments in Streptomyces cacaoi demonstrated that PolF is absolutely essential for polyoxin A production (<1% of wild-type levels), while PolE disruption resulted in substantially reduced titers (~10% of wild-type) [5] [10]. This genetic evidence established PolF as the central catalyst in azetidine formation, with PolF playing an auxiliary role in enhancing pathway efficiency.
Table 1: Key Enzymes in Azetidine Amino Acid Biosynthesis
| Enzyme | Family | Cofactor Requirement | Catalytic Function | Essentiality |
|---|---|---|---|---|
| PolF | Heme-oxygenase-like dimetal oxidase/oxygenase (HDO) | Diiron center (Feâ‚‚) | Transforms l-Ile and l-Val to azetidine derivatives via desaturated intermediate | Essential (<1% product without PolF) |
| PolE | DUF6421 | Fe and pterin | Catalyzes desaturation of l-Ile, increasing pathway flux | Non-essential but increases titer (~10% product without PolE) |
Enzymatic Mechanisms of Azetidine Formation
PolF: A Novel HDO Enzyme Catalyzing Azetidine Ring Formation
PolF represents a newly discovered member of the heme-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily, an emerging class of diiron-dependent enzymes that activate Oâ‚‚ for diverse oxidative transformations [5] [10]. Structural homology analysis using Foldseek revealed that PolF shares structural features with characterized HDO enzymes, despite lacking significant sequence similarity [5]. Notably, PolF expands the catalytic repertoire of HDO enzymes, as no previously characterized family member was known to construct azetidine rings.
Biochemical characterization demonstrated that PolF is sufficient to convert l-Ile to polyoximic acid in vitro when reconstituted with Fe(II) under aerobic conditions [5]. The enzyme exhibits a strict metal requirement, with activity observed only with Fe(II) among tested transition metals [5]. Activity dependence on external reductants such as ascorbate or dithiothreitol aligns with the established need for reducing equivalents during multiple catalytic turnovers of HDO enzymes [5] [10].
Table 2: Substrate Specificity of PolF
| Substrate | Major Product(s) | Structural Requirement | Proposed Rationale |
|---|---|---|---|
| l-Isoleucine | Polyoximic acid (azetidine) | β-Methyl group | Steric and electronic stabilization of radical intermediate |
| l-Valine | 3-methylene-azetidine-2-carboxylic acid (MAA) | β-Methyl group | Similar branching to l-Ile at β-position |
| l-Leucine | Hydroxylation products (minor desaturation) | Lack of β-methyl group | Altered radical migration pathway |
| l-Methionine | Hydroxylation products (minor desaturation) | Sulfur-containing side chain | Competing reaction pathways |
| Other proteogenic amino acids | No detectable products | - | Specific active site constraints |
Mechanistic Insights into PolF Catalysis
Detailed investigation of PolF catalysis using l-valine as a substrate revealed a complex reaction mechanism involving multiple intermediates [5]. Under single-turnover conditions, the following species were detected, indicating a branched catalytic pathway:
- 3,4-dehydrovaline (3,4-dh-Val): The major initial product, characterized by a -2 Da mass shift corresponding to desaturation.
- 3-dimethylaziridine-2-carboxylic acid (Azi): A three-membered ring intermediate demonstrating PolF's capacity for C–N bond formation.
- 4-hydroxyvaline and 3-hydroxyvaline: Hydroxylation products representing alternative oxidative outcomes.
The conversion of 3,4-dh-Val to the azetidine product MAA under both enzymatic and chemical conditions suggests that the desaturated intermediate can undergo non-enzymatic cyclization, though the enzymatic pathway significantly accelerates this process [5].
Spectroscopic and structural studies indicate that PolF employs a μ-peroxo-Fe(III)â‚‚ species to initiate catalysis by cleaving an unactivated C–H bond with a bond dissociation energy up to 101 kcal molâ»Â¹ [5] [10]. This potent oxidant abstracts a hydrogen atom from the substrate, generating a carbon-centered radical that subsequently participates in C–N bond formation through radical mechanisms [5]. Crystal structures of PolF in complex with l-Ile reveal precise substrate positioning that orients the reactive centers for selective azetidine formation over alternative oxidative outcomes.
The following diagram illustrates the proposed catalytic cycle of PolF:
PolE: A Supplementary Enzyme Enhancing Biosynthetic Flux
PolE, a member of the DUF6421 family, functions as an Fe- and pterin-dependent oxidase that catalyzes the desaturation of l-Ile to 3,4-dehydroisoleucine [5] [10]. This activity positions PolE as an auxiliary enzyme that increases the local concentration of the desaturated intermediate, thereby enhancing the flux through the azetidine biosynthetic pathway. The coordinated action of PolE and PolF represents an efficient metabolic strategy for producing the strained azetidine ring system.
Experimental Characterization of Azetidine Biosynthesis
Gene Knockout and Microbial Fermentation
Protocol: Gene Disruption in Streptomyces cacaoi
- Strain Preparation: Culture wild-type S. cacaoi under polyoxin-producing conditions.
- Gene Deletion: Perform in-frame deletion of polE and polF genes using standard genetic techniques.
- Fermentation: Culture wild-type and mutant strains under identical polyoxin-producing conditions.
- Metabolite Extraction: Harvest fermentation broth and extract metabolites using appropriate solvents.
- LC-MS Analysis: Analyze polyoxin A production using liquid chromatography-mass spectrometry with multiple reaction monitoring.
- Quantification: Compare peak areas of polyoxin A in mutants versus wild-type strains.
This genetic approach established that PolF is absolutely essential for polyoxin biosynthesis, while PolE significantly enhances production yield [5] [10].
In Vitro Enzyme Assays
Protocol: PolF Activity Assay
- Enzyme Preparation: Express and purify PolF from E. coli in predominantly apo form.
- Metallation: Incubate apo-PolF with 3 equivalents of Fe(II) under anaerobic conditions in an inert atmosphere chamber.
- Excess Metal Removal: Remove unbound Fe(II) using a desalting column.
- Reaction Setup: Prepare enzyme-substrate mixture anaerobically with l-Ile or l-Val as substrate.
- Reaction Initiation: Inject Oâ‚‚-saturated buffer to initiate the oxidative reaction.
- Product Derivatization: Treat reaction products with dansyl chloride (DnsCl) for enhanced detection.
- Analysis: Identify and quantify products using LC-MS with comparison to authentic standards.
Critical controls include reactions without Fe(II), without Oâ‚‚, and with heat-inactivated enzyme, all of which should show no product formation [5].
Intermediate Trapping and Characterization
Protocol: Single-Turnover Experiments
- Enzyme Preparation: Reconstitute PolF with precisely 2 equivalents of Fe(II).
- Reaction Setup: Combine enzyme and substrate under anaerobic conditions without external reductant.
- Rapid Mixing: Expose to Oâ‚‚ for defined time intervals.
- Reaction Quenching: Acidify or flash-freeze to terminate reactions at specific timepoints.
- Intermediate Identification: Analyze timepoints by LC-MS to detect transient intermediates.
- Kinetic Analysis: Determine relative rates of formation for different intermediates.
This approach identified 3,4-dh-Val as the major initial product, followed by Azi, 4-OH-Val, and 3-OH-Val, providing critical insights into the branched reaction mechanism [5].
The following workflow summarizes the key experimental approaches for characterizing azetidine biosynthesis:
Research Reagent Solutions
Table 3: Essential Research Reagents for Studying Azetidine Biosynthesis
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Enzyme Sources | Recombinant PolF and PolE expressed in E. coli | Catalytic core of azetidine formation; study structure-function relationships |
| Metal Cofactors | Fe(II) salts (e.g., FeSOâ‚„) | Essential cofactor for both PolF and PolE; reconstitutes active enzymes |
| Substrates | l-Isoleucine, l-Valine, l-allo-Isoleucine | Natural substrates for azetidine formation; study substrate specificity |
| Analytical Standards | Polyoximic acid (from polyoxin A hydrolysis), 3,4-dehydrovaline | Reference compounds for product identification and quantification |
| Derivatization Reagents | Dansyl chloride (DnsCl) | Enhances detection sensitivity of amino acid products in LC-MS |
| Reducing Agents | Ascorbate, dithiothreitol (DTT) | Maintains Fe in reduced state during multiple turnover experiments |
| Chromatography | Reverse-phase LC columns, mobile phases | Separation and analysis of substrates, intermediates, and products |
Implications and Future Perspectives
The discovery of PolF and PolE as the catalysts for azetidine amino acid biosynthesis represents a significant advancement in natural product biochemistry. This pathway demonstrates nature's ingenuity in employing radical chemistry to construct strained ring systems that challenge conventional synthetic approaches. The identification of PolF as a novel HDO enzyme expands the functional diversity of this emerging superfamily and provides new insights into diiron-mediated C–H activation [5] [10].
From a pharmaceutical perspective, understanding this biosynthetic route enables potential bioengineering approaches to produce azetidine-containing compounds with enhanced properties. The azetidine ring's capacity to modulate molecular rigidity, improve metabolic stability, and enhance target binding affinity makes it particularly valuable in drug design [7]. The enzymatic machinery revealed in this pathway offers potential biocatalytic tools for sustainable synthesis of these valuable motifs under mild conditions, contrasting with traditional chemical methods that often require harsh reagents and elevated temperatures [7].
Future research directions include detailed structural characterization of the enzyme-substrate complexes, exploration of the enzyme's engineering potential for producing azetidine derivatives, and investigation of whether similar mechanisms operate in other azetidine-containing natural products. The interplay between PolE and PolF also presents an interesting model for studying metabolic channeling and pathway optimization in natural product biosynthesis.
The azetidine ring, a four-membered nitrogen-containing saturated heterocycle, is a crucial structural motif in numerous bioactive compounds and pharmaceuticals [5] [10]. Its high ring strain of approximately 25.4 kcal molâ»Â¹ makes it a valuable substrate for various chemical transformations, including ring opening, expansion, and metal-catalyzed reactions [5] [10]. Despite its significance, the enzymatic pathways responsible for azetidine biosynthesis have remained largely enigmatic, creating a major bottleneck in understanding natural product biosynthesis and developing biocatalytic applications [5] [10] [11].
Historically, researchers have identified only a limited number of enzymatic mechanisms capable of forming this strained ring system. The two principal known pathways—S-adenosyl-L-methionine (SAM)-dependent cyclization and α-ketoglutarate (α-KG)/Fe-dependent oxygenase catalysis—have significant biochemical and practical limitations that restrict their utility in drug discovery and bioengineering [5] [10] [11]. These pathways require metabolically expensive precursors, exhibit limited substrate scope, and offer minimal flexibility for synthetic biology applications.
This review comprehensively analyzes the historical constraints of established azetidine biosynthetic pathways, providing a detailed examination of their mechanistic limitations and biochemical requirements. Furthermore, we contextualize these limitations within the recent groundbreaking discovery of a new biosynthetic route mediated by non-haem iron-dependent enzymes PolE and PolF in the polyoxin antifungal pathway [5] [10] [11]. By framing this new pathway as a solution to longstanding challenges, this review provides researchers with a comprehensive understanding of the evolving landscape of azetidine biosynthesis.
Established Azetidine Biosynthetic Pathways and Their Constraints
SAM-Dependent Azetidine Formation
The best-characterized mechanism for azetidine ring formation involves S-adenosyl-L-methionine (SAM)-dependent enzymes that catalyze an intramolecular nucleophilic cyclization [5] [10]. In this pathway, SAM serves as both the substrate and cofactor, undergoing an internal cyclization to yield azetidine carboxylic acid and 5'-methylthioadenosine (MTA) as a byproduct [10].
Table 1: Limitations of SAM-Dependent Azetidine Biosynthesis
| Limitation Factor | Biochemical Impact | Practical Consequence |
|---|---|---|
| Metabolically Expensive Precursor | Requires SAM, consuming ATP equivalents | Energetically costly for microbial production systems |
| Fixed Product Structure | Produces only azetidine-2-carboxylic acid | Limited chemical diversity for drug discovery |
| Byproduct Accumulation | Generates 5'-methylthioadenosine (MTA) | Potential feedback inhibition and metabolic burden |
| Limited Substrate Scope | Restricted to SAM or close analogs | Difficult to generate azetidine derivatives with side-chain variations |
The SAM-dependent pathway presents substantial metabolic challenges for industrial or biocatalytic applications. The requirement for SAM, an expensive nucleotide derivative, increases the metabolic burden on production hosts [5]. Furthermore, the structural limitation of producing exclusively azetidine-2-carboxylic acid constrains its utility for generating diverse azetidine-containing compounds with varied pharmaceutical properties [5] [10].
α-Ketoglutarate and Fe-Dependent Oxygenase Pathway
An alternative azetidine formation mechanism occurs in the biosynthesis of okaramine, where an α-ketoglutarate (α-KG) and Fe-dependent oxygenase catalyzes a radical-mediated oxidative C–C bond formation to produce the azetidine ring [5] [10] [11]. This pathway represents a distinct biochemical strategy that diverges from the SAM-dependent cyclization mechanism.
Table 2: Limitations of α-KG/Fe-Dependent Azetidine Biosynthesis
| Limitation Factor | Biochemical Impact | Practical Consequence |
|---|---|---|
| Cofactor Requirement | Needs α-ketoglutarate as essential cosubstrate | Increased metabolic cost and complexity |
| Stoichiometric Byproduct | Forms succinate and COâ‚‚ for each reaction cycle | Potential metabolic imbalance in production hosts |
| Radical Intermediate Control | Requires precise radical stabilization | Limited enzyme engineering potential |
| Substrate Specificity | Highly specific to native biosynthetic context | Difficult to repurpose for novel compound production |
The α-KG-dependent pathway, while mechanistically distinct from the SAM-dependent route, shares similar practical limitations [5]. The requirement for α-ketoglutarate as an essential cosubstrate adds significant metabolic cost, while the production of stoichiometric byproducts (succinate and CO₂) complicates bioprocess optimization [5] [11]. Additionally, the need for precise control of radical intermediates limits engineering possibilities for creating modified azetidine structures.
Experimental Characterization of Azetidine Biosynthesis
Gene Inactivation Studies
Critical insights into azetidine biosynthesis emerged from targeted gene knockout experiments in the polyoxin producer Streptomyces cacaoi [5] [10]. Researchers performed in-frame deletion of polE and polF genes, then cultured the mutants under polyoxin-producing conditions. The fermentation broth was analyzed using liquid chromatography-mass spectrometry (LC-MS) to quantify polyoxin production [5] [10].
Protocol: Gene Knockout and Metabolite Analysis
- In-frame gene deletion: Precisely delete polE and polF genes from S. cacaoi genome (Supplementary Fig. 1 [5])
- Fermentation culture: Grow wild-type and mutant strains under polyoxin-producing conditions
- Metabolite extraction: Process fermentation broth for LC-MS analysis
- LC-MS analysis: Quantify polyoxin A production using validated methods
- Data interpretation: Compare metabolite profiles between strains
The gene inactivation results demonstrated that PolF is absolutely essential for polyoximic acid biosynthesis, with the polF mutant producing less than 1% of wild-type polyoxin A levels [5] [10]. In contrast, the polE mutant produced approximately 10% of wild-type levels, indicating a supplementary rather than essential role for PolE [5] [10]. These findings established PolF as the core enzymatic component responsible for azetidine formation.
Enzyme Expression and Purification
To biochemically characterize PolF, researchers expressed and purified the enzyme from E. coli (Supplementary Fig. 3 [5] [10]). The initial purification yielded predominantly apo-PolF, consistent with the weak iron affinity reported for haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily enzymes in the absence of substrate [5] [10].
Protocol: PolF Reconstitution and Activity Assay
- Enzyme purification: Express PolF in E. coli and purify using affinity chromatography
- Iron reconstitution: Incubate apo-PolF with 3 equivalents of Fe(II) under anaerobic conditions
- Excess iron removal: Use desalting column to remove unbound Fe(II)
- Iron quantification: Measure iron content (typically ~1.5 eq. per PolF) [5] [10]
- Anaerobic assay preparation: Mix enzyme and substrate in oxygen-free environment
- Reaction initiation: Add Oâ‚‚-saturated buffer to start reaction
- Product derivatization: Treat reaction products with dansyl chloride (DnsCl)
- Product analysis: Analyze derivatized products by LC-MS (Supplementary Figs. 4-6 [5])
Activity assays demonstrated that PolF exclusively catalyzes the transformation of L-isoleucine to polyoximic acid when reconstituted with Fe(II) and oxygen [5] [10]. Control reactions lacking Fe(II), Oâ‚‚, or containing heat-inactivated PolF showed no product formation, confirming the enzyme's dependence on both iron and oxygen [5].
Substrate Specificity Profiling
Comprehensive substrate specificity studies revealed that PolF reacts selectively with medium-size aliphatic amino acids [5] [10]. When tested against the 20 proteogenic amino acids, PolF showed significant activity toward L-valine, producing 3-methylene-azetidine-2-carboxylic acid (MAA) as a -4 Da modified product (Supplementary Fig. 9 [5] [10]). L-leucine and L-methionine yielded primarily hydroxylation products with minor desaturation products, while other amino acids produced no detectable products [5] [10].
Table 3: PolF Substrate Specificity and Products
| Substrate | Major Product(s) | Structural Requirement |
|---|---|---|
| L-isoleucine | Polyoximic acid (PA) | β-methyl group critical |
| L-valine | 3-methylene-azetidine-2-carboxylic acid (MAA) | Medium-size aliphatic chain |
| L-leucine | Hydroxylation products (minor desaturation) | Branched chain but no azetidine |
| L-methionine | Hydroxylation products (minor desaturation) | Sulfur-containing but no azetidine |
| Other proteogenic amino acids | No detectable products | Specificity for aliphatic substrates |
Stereochemical studies using L-isoleucine stereoisomers revealed that while L-allo-Ile, D-Ile, and D-allo-Ile all yielded small amounts of azetidine products, the stereochemistries at C2 and C3 positions significantly influence catalytic efficiency [5] [10].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Azetidine Biosynthesis Studies
| Reagent / Material | Function / Application | Experimental Context |
|---|---|---|
| Apo-PolF enzyme | Catalytic core for azetidine formation | Recombinantly expressed in E. coli [5] [10] |
| Fe(II) solution | Cofactor for enzyme reconstitution | Anaerobic preparation essential [5] [10] |
| Oâ‚‚-saturated buffer | Oxidant for initiating catalysis | Reaction initialization [5] [10] |
| Ascorbate/DTT | External reductant for multiple turnovers | Maintains diiron center reduction state (Supplementary Fig. 7 [5]) |
| Dansyl chloride (DnsCl) | Derivatization agent for LC-MS detection | Enables product visualization and quantification [5] [10] |
| L-Ile, L-Val analogs | Substrate specificity mapping | Comprehensive activity profiling [5] [10] |
| Polyoxin A standard | Authentic standard for PA identification | Product verification (Supplementary Fig. 5 [5]) |
| Boc-L-Pra-OH (DCHA) | N-cyclohexylcyclohexanamine;(2S)-2-[(2-methylpropan-2-yl)oxycarbonylamino]pent-4-ynoic acid | This product, N-cyclohexylcyclohexanamine;(2S)-2-[(2-methylpropan-2-yl)oxycarbonylamino]pent-4-ynoic acid (CAS 63039-49-6), is a dicyclohexylamine (DCHA) salt of a Boc-protected amino acid for research use only (RUO). It is not for personal, veterinary, or household use. |
| Methyl (tert-butoxycarbonyl)-L-leucinate | Methyl (tert-butoxycarbonyl)-L-leucinate, CAS:63096-02-6, MF:C12H23NO4, MW:245.32 g/mol | Chemical Reagent |
Mechanistic Workflow and Pathway Relationships
The experimental characterization of azetidine biosynthesis involves a logical progression from genetic validation to biochemical mechanism elucidation. The following diagram maps this workflow and the relationships between key experimental approaches:
Transition to a New Biosynthetic Paradigm
The historical limitations of SAM-dependent and α-KG-dependent pathways created a compelling need for alternative azetidine biosynthesis mechanisms. The constraints of these established pathways—particularly their metabolic costs, limited product diversity, and restricted substrate scope—hindered progress in both understanding natural product biosynthesis and developing biocatalytic applications [5] [10] [11].
The recent discovery of the non-haem iron-dependent pathway featuring PolF represents a significant paradigm shift in azetidine biosynthesis [5] [10] [11]. This new mechanism operates through a fundamentally different biochemical strategy, utilizing a μ-peroxo-Fe(III)₂ intermediate for unactivated C–H bond cleavage and proceeding through radical mechanisms for C–N bond formation [5] [10]. Unlike previous pathways, the PolF system transforms simple, readily available amino acid precursors (L-Ile, L-Val) into azetidine derivatives via 3,4-desaturated intermediates, bypassing the requirement for expensive cofactors [5] [10].
This breakthrough not only addresses the historical limitations outlined in this review but also substantially expands the toolbox available for synthetic biology and drug development. The mechanistic insights from PolF catalysis, combined with the auxiliary function of PolE in enhancing desaturation flux, provide a new foundation for engineering azetidine biosynthesis in heterologous systems and developing novel biocatalytic applications for pharmaceutical development [5] [7] [10].
The azetidine ring, a strained four-membered nitrogen-containing heterocycle, is a crucial structural feature in numerous bioactive compounds and pharmaceuticals [5] [11]. Its high ring strain (approximately 25.4 kcal molâ»Â¹) makes it both challenging to synthesize and a valuable scaffold in drug discovery [5]. For decades, the biosynthetic pathways leading to this ring in natural products remained largely enigmatic. Prior to the discovery of the Polyoxin system, only two known enzymatic mechanisms for azetidine formation were characterized: (1) S-adenosyl-L-methionine (SAM)-dependent enzymes that catalyze an intramolecular nucleophilic cyclization, and (2) α-ketoglutarate (α-KG) and Fe-dependent oxygenases that facilitate radical-mediated oxidative C–C bond formation [5] [11]. Both these routes depend on metabolically expensive precursors, limiting their utility for biocatalytic applications [11].
The antifungal nucleoside polyoxin A, produced by Streptomyces cacaoi, contains an azetidine amino acid known as polyoximic acid (PA) [5] [12]. Early isotope-labelling studies indicated that PA is derived from L-isoleucine (L-Ile), suggesting the existence of a novel biosynthetic mechanism distinct from the known pathways [5]. Within the polyoxin biosynthetic gene cluster, two genes coding for putative enzymes, PolE and PolF, were identified as likely participants in the construction of the azetidine ring, yet their precise functions were unknown [5] [11] [13]. This guide details the elucidation of this novel pathway, focusing on the groundbreaking discovery of the non-haem iron-dependent enzymes PolE and PolF.
The Polyoxin System: Gene Cluster and Essential Enzymes
The polyoxin biosynthetic gene cluster (BGC0000877) from Streptomyces cacaoi subsp. asoensis contains the genes polE and polF, which were initially annotated as a hypothetical protein and a putative molybdopterin oxidoreductase, respectively [13]. Functional analysis through gene knockout experiments established their critical roles. Disruption of the polF gene completely abolished polyoxin A production (<1% of wild-type), demonstrating that PolF is essential for PA biosynthesis. In contrast, a polE mutant produced a reduced but detectable amount of polyoxin A (~10% of wild-type), indicating that PolE plays a supporting role in enhancing the biosynthetic flux [5].
Table 1: Key Enzymes in the Polyoxin Azetidine Biosynthetic Pathway
| Enzyme | Gene Locus | Protein Family | Cofactors | Essential for Production? | Primary Function |
|---|---|---|---|---|---|
| PolF | ABX24498.1 [13] | HDO (Haem-oxygenase-like dimetal oxidase/or oxygenase) superfamily [5] | Diiron (Feâ‚‚) [5] | Yes (<1% yield without polF) [5] | Catalyzes azetidine ring formation from L-Ile and L-Val [5] |
| PolE | ABX24499.1 [13] | DUF6421 family [5] | Iron (Fe) and pterin [5] | No (~10% yield without polE) [5] | Assists PolF by catalyzing desaturation of L-Ile [5] |
Functional Characterization of the Key Enzyme PolF
PolF is an HDO Superfamily Enzyme
Bioinformatic analysis revealed that PolF is a member of the haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily, an emerging group of diiron-dependent enzymes that activate Oâ‚‚ to perform diverse oxidative reactions [5]. Unlike previously characterized HDOs, PolF possesses the unique ability to construct an azetidine ring.
In Vitro Reconstitution of PolF Activity
To confirm its function, PolF was heterologously expressed in E. coli and purified. The enzyme was reconstituted with Fe(II) under anaerobic conditions for functional assays [5].
Table 2: Summary of PolF Activity Assay Conditions and Results
| Assay Component | Condition | Observation | Conclusion |
|---|---|---|---|
| Enzyme | Active PolF | PA production detected | PolF is necessary for reaction [5] |
| Heat-inactivated PolF | No PA detected | ||
| Cofactor | With Fe(II) | PA production detected | Fe(II) is essential metal cofactor [5] |
| With other transition metals (e.g., Mn, Co, Ni) | No PA detected | ||
| Atmosphere | Aerobic (Oâ‚‚ present) | PA production detected | Oâ‚‚ is essential co-substrate [5] |
| Anaerobic | No PA detected | ||
| Reductant | With ascorbate or DTT | Multiple turnovers observed | External reductant required for catalytic cycling [5] |
| No external reductant | Limited turnover |
The assay mixture, containing L-Ile and Fe-reconstituted PolF, was initiated by introducing an Oâ‚‚-saturated buffer. Subsequent LC-MS analysis of the dansyl chloride-derivatized products revealed a compound with a mass 4 Da less than L-Ile, which was identified as polyoximic acid (PA) by comparison with an authentic standard from polyoxin A [5].
Substrate Specificity and Catalytic Versatility of PolF
Investigation into PolF's substrate scope revealed its ability to accept L-valine (L-Val) as a substrate, converting it to 3-methylene-azetidine-2-carboxylic acid (MAA). Among other proteogenic amino acids, only L-leucine and L-methionine yielded minor products, primarily hydroxylated derivatives, but no azetidine rings, highlighting a specificity for medium-sized aliphatic amino acids and suggesting the β-methyl group is critical for cyclization [5].
Under single-turnover conditions, PolF was found to catalyze three distinct reactions on L-Val: desaturation (producing 3,4-dehydrovaline, 3,4-dh-Val), hydroxylation (producing 3- and 4-hydroxyvaline), and C–N bond formation (producing an azetidine-containing product and an intermediate aziridine, 3-dimethylaziridine-2-carboxylic acid). This demonstrates the remarkable catalytic versatility of a single HDO enzyme [5].
The Supporting Role of PolE
Functional characterization showed that PolE, a member of the DUF6421 family, is an Fe and pterin-dependent oxidase [5]. It catalyzes the desaturation of L-Ile to a 3,4-desaturated intermediate. This activity primes the substrate for PolF, increasing the efficiency and specificity of the pathway by providing a more direct flux toward the azetidine ring compared to the multi-functional catalysis of PolF alone [5].
Proposed Biosynthetic Pathway and Mechanism
Based on genetic, enzymological, and structural data, the biosynthetic route to the azetidine ring in polyoximic acid can be summarized as follows.
Diagram 1: Biosynthetic pathway to polyoximic acid. PolE and PolF can act sequentially or PolF can act alone on L-Ile.
The mechanism of PolF involves a μ-peroxo-Fe(III)₂ intermediate that is directly responsible for the challenging cleavage of the unactivated C–H bond [5]. Subsequent steps, including the crucial C–N bond formation, are proposed to proceed through radical mechanisms [5] [11]. The crystal structure of PolF in complex with L-Ile has provided critical insights into how the enzyme facilitates this strained ring cyclization [5].
Experimental Protocols for Key Assays
Enzyme Purification and Reconstitution
- Heterologous Expression: Express PolF in E. coli and purify using standard chromatographic techniques (e.g., affinity and size-exclusion chromatography) [5].
- Apo-Enzyme Preparation: Isolate PolF in its apo-form, largely devoid of metal cofactors [5].
- Anaerobic Reconstitution: Incubate apo-PolF with a 3-fold molar excess of Fe(II) under strictly anaerobic conditions (e.g., in a glovebox or using Schlenk techniques) [5].
- Removal of Excess Metal: Pass the reconstituted mixture through a desalting column (e.g., PD-10) to remove unbound Fe(II). Typical reconstitution yields PolF with ~1.5 equivalents of bound Fe(II), consistent with the weak metal affinity of HDO enzymes in the absence of substrate [5].
PolF Activity Assay
- Reaction Setup: Prepare an anaerobic mixture containing the reconstituted PolF (~1.5 eq Fe) and substrate (e.g., 1 mM L-Ile or L-Val) in a suitable buffer [5].
- Initiate Reaction: Start the reaction by rapidly adding an Oâ‚‚-saturated buffer to the mixture. Include controls without enzyme, without Fe(II), without Oâ‚‚, and with heat-inactivated enzyme [5].
- Include Reductant: For multiple turnover assays, include an external reductant like ascorbate or dithiothreitol (DTT) in the reaction mixture to re-reduce the diiron centre [5].
- Product Derivatization and Analysis:
- Derivatize reaction products with dansyl chloride (DnsCl) [5].
- Analyze derivatized products using Liquid Chromatography-Mass Spectrometry (LC-MS) [5].
- Identify polyoximic acid by comparison of retention time and mass with an authentic standard. A product with a mass of -4 Da relative to the substrate indicates the formation of the azetidine ring [5].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Studying the Polyoxin Azetidine Pathway
| Reagent / Material | Function in Research | Key Details / Examples |
|---|---|---|
| Apo-PolF Enzyme | Catalytic core for in vitro mechanistic studies. | Heterologously expressed and purified from E. coli [5]. |
| Fe(II) Salts | Essential metallo-cofactor for reconstituting active PolF and PolE. | e.g., Ammonium iron(II) sulfate or FeSOâ‚„; handling requires anaerobic conditions [5]. |
| L-Isoleucine (L-Ile) | Native biosynthetic substrate. | Used in activity assays and kinetic studies [5]. |
| L-Valine (L-Val) | Alternative substrate for probing enzyme mechanism and specificity. | Yields 3-methylene-azetidine-2-carboxylic acid (MAA) [5]. |
| Oâ‚‚-saturated Buffer | Co-substrate for the oxidative reaction. | Prepared by bubbling oxygen through the buffer immediately before assay initiation [5]. |
| External Reductants | Maintains catalytic cycling during multiple turnovers. | e.g., Ascorbate or Dithiothreitol (DTT) [5]. |
| Dansyl Chloride (DnsCl) | Derivatization agent for enhancing LC-MS detection of amino acid substrates and products. | Reacts with amine groups for sensitive fluorescence and MS detection [5]. |
| Authentic PA Standard | Reference for confirming product identity in chromatographic analyses. | Can be prepared from hydrolyzed polyoxin A [5]. |
| Boc-L-Ala-OH | Boc-L-Ala-OH, CAS:15761-38-3, MF:C8H15NO4, MW:189.21 g/mol | Chemical Reagent |
| Dabcyl acid | Dabcyl acid, CAS:6268-49-1, MF:C15H15N3O2, MW:269.30 g/mol | Chemical Reagent |
The discovery of the Polyoxin system, centered on the enzymes PolE and PolF, represents a paradigm shift in our understanding of azetidine biosynthesis. This pathway unveils a third, distinct mechanistic strategy for forming the strained azetidine ring, fundamentally different from SAM-dependent cyclization or α-KG/Fe-dependent oxygenase mechanisms [5] [11]. The characterization of PolF as a diiron-dependent HDO enzyme capable of catalyzing desaturation, hydroxylation, and radical-mediated C–N bond formation significantly expands the known catalytic repertoire of this enzyme superfamily [5].
From a biotechnological perspective, this novel pathway is particularly promising. Unlike previous routes, it utilizes readily available proteinogenic amino acids (L-Ile, L-Val) as starting materials, bypassing the need for expensive precursors and potentially enabling more efficient biocatalytic applications for the synthesis of azetidine-containing pharmaceuticals and fine chemicals [11]. Future research will likely focus on further elucidating the detailed radical rearrangement mechanism, engineering PolF for altered substrate scope and enhanced stability, and exploring its application in synthetic biology for the production of valuable azetidine compounds.
Azetidine, a saturated four-membered nitrogen-containing heterocycle, is a crucial structural motif in numerous bioactive compounds and drugs [5] [11]. Its high ring strain (approximately 25.4 kcal molâ»Â¹) makes it both energetically challenging to form and a valuable substrate for various chemical transformations [5]. In nature, the azetidine ring is found in important molecules such as polyoximic acid (PA), the azetidine amino acid present in the antifungal nucleoside polyoxin A [5] [11]. Early isotope-labelling studies indicated that PA is derived from L-isoleucine (L-Ile), suggesting a biosynthetic pathway distinct from previously known mechanisms [5].
Until recently, the characterized enzymatic mechanisms for azetidine ring formation were limited. The best-known pathways involve S-adenosyl-L-methionine (SAM)-dependent enzymes that catalyze an intramolecular nucleophilic cyclization, or α-ketoglutarate (α-KG)- and Fe-dependent oxygenases that mediate a radical-based oxidative C–C bond formation [5] [11]. A significant limitation of these routes is their dependence on metabolically or chemically expensive precursors, which restricts their utility for biocatalytic applications [11]. The biosynthesis of the azetidine ring in polyoxin, therefore, presented an intriguing enigma. Within the polyoxin biosynthetic gene cluster, two genes encoding putative enzymes, PolE and PolF, were proposed to be involved in PA biosynthesis, but their specific functions and mechanisms remained unknown [5] [11]. This whitepaper details the elucidation of PolF as a novel non-haem iron-dependent enzyme from the haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily, responsible for the unique transformation of L-amino acids into azetidine derivatives.
PolF: Essential Role and Catalytic Function
Genetic Evidence for PolF's Essential Role
The critical role of polF in azetidine biosynthesis was unequivocally demonstrated through targeted gene knockout experiments in Streptomyces cacaoi, the native polyoxin producer [5]. Analysis of the fermentation broth using liquid chromatography–mass spectrometry (LC–MS) revealed that inactivation of polF completely abolished the production of polyoxin A, reducing titers to below 1% of wild-type levels [5]. In contrast, a polE mutant still produced detectable amounts of polyoxin A, albeit at a significantly reduced yield (~10% of wild-type) [5]. These genetic findings established that PolF is indispensable for polyoximic acid biosynthesis, while PolE appears to play a supporting, non-essential role in enhancing biosynthetic flux.
In Vitro Characterization of PolF Activity
Recombinant PolF was expressed and purified from E. coli in its apo-form [5]. Functional characterization confirmed its activity as a diiron-dependent enzyme. Table 1 summarizes the key experimental parameters and conditions for the in vitro reconstitution of PolF activity.
Table 1: Summary of PolF Activity Assay Conditions
| Parameter | Description/Value | Function/Note |
|---|---|---|
| Enzyme Form | Apo-PolF | Purified from E. coli; requires Fe(II) reconstitution |
| Metal Cofactor | Fe(II) | Essential; other transition metals (e.g., Mn, Co, Ni, Cu) could not substitute [5] |
| Reconstitution | Incubation with 3 eq. Fe(II) under anaerobic conditions | Resulted in ~1.5 eq. Fe bound per PolF, consistent with weak Fe affinity in HDOs without substrate [5] |
| Reaction Initiation | Addition of Oâ‚‚-saturated buffer | Requires molecular oxygen |
| External Reductant | Ascorbate or Dithiothreitol (DTT) | Required for multiple turnovers to re-reduce the diiron center [5] |
| Product Detection | Derivatization with dansyl chloride (DnsCl) followed by LC-MS | Enabled detection and identification of reaction products |
Assays performed under these conditions, with L-Ile as the substrate, successfully produced a compound with a molecular mass 4 Da lower than L-Ile [5]. This product was confirmed to be polyoximic acid (PA) by comparison with an authentic standard derived from polyoxin A and through structural characterization by NMR [5]. Control reactions lacking Fe(II), Oâ‚‚, or using heat-inactivated PolF yielded no PA, confirming the enzyme's specific and catalytic role in this transformation [5].
Catalytic Mechanism and Substrate Specificity
Proposed Catalytic Cycle of PolF
PolF is a member of the haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily, an emerging class of diiron enzymes that activate Oâ‚‚ for diverse oxidative reactions [5]. Mechanistic studies, including spectroscopic characterization, suggest that a μ-peroxo-Fe(III)â‚‚ intermediate is directly responsible for the initial, chemically challenging cleavage of an unactivated C–H bond (bond strength up to 101 kcal molâ»Â¹) [5]. Subsequent steps, including the critical C–N bond formation that closes the azetidine ring, are proposed to proceed via radical mechanisms [5]. The following diagram illustrates the current understanding of the PolF catalytic cycle, integrating the formation of key intermediates.
Diagram 1: Proposed catalytic cycle of PolF, highlighting the key μ-peroxo-Fe(III)₂ intermediate and radical-mediated C-N bond formation.
Identification of Intermediates and Reaction Landscape
The catalytic versatility of PolF was revealed through experiments using L-valine (L-Val) as a substrate, which allowed for the detection and characterization of several intermediates [5]. Under multiple turnover conditions, the primary product from L-Val is 3-methyene-azetidine-2-carboxylic acid (MAA) [5]. However, under single-turnover conditions (using 2 equivalents of Fe(II) and no external reductant), several intermediates were observed, indicating that PolF can catalyze three distinct reactions on a single substrate [5]:
- Desaturation: Formation of 3,4-dehydrovaline (3,4-dh-Val), the major initial product.
- Hydroxylation: Formation of 3-hydroxyvaline (3-OH-Val) and 4-hydroxyvaline (4-OH-Val).
- C–N Bond Formation: Formation of 3-dimethylaziridine-2-carboxylic acid (Azi), a direct precursor to the azetidine ring.
Notably, when 3,4-dh-Val was used as a substrate, it was quantitatively converted to MAA, identifying it as a key intermediate en route to the azetidine product [5]. This suggests a pathway where desaturation precedes azetidine ring closure.
Substrate Scope and Specificity
The substrate specificity of PolF was probed using the 20 proteogenic amino acids. Table 2 summarizes the reactivity of different amino acid substrates with PolF, highlighting its selectivity for medium-sized aliphatic amino acids.
Table 2: Substrate Specificity of PolF
| Substrate | Product(s) | Relative Reactivity / Notes |
|---|---|---|
| L-Isoleucine (L-Ile) | Polyoximic Acid (PA) | Native substrate; azetidine ring formed efficiently [5] |
| L-Valine (L-Val) | 3-Methylene-azetidine-2-carboxylic acid (MAA) | Azetidine ring formed efficiently [5] |
| L-Leucine (L-Leu) | Mostly hydroxylation products; minor desaturation | No azetidine product detected [5] |
| L-Methionine (L-Met) | Mostly hydroxylation products; minor desaturation | No azetidine product detected [5] |
| Other Proteogenic Amino Acids | No detectable products | PolF reacts selectively with medium-size aliphatic amino acids [5] |
| L-allo-Ile, D-Ile, D-allo-Ile | Small amounts of azetidine products | Stereochemistry at C2 and C3 is important but not absolutely essential [5] |
The data indicate that PolF exhibits a strict requirement for a β-methyl group for successful azetidine formation, as neither L-Leu nor L-Met yielded azetidine products [5]. The enzyme also shows a degree of stereochemical flexibility, as all stereoisomers of isoleucine were transformed into azetidine products, albeit less efficiently [5].
The Supporting Role of PolE
While PolF alone is sufficient to convert L-Ile to PA, the biosynthetic pathway is assisted by PolE, a member of the DUF6421 family [5] [11]. Functional characterization revealed that PolE is an iron and pterin-dependent oxidase that specifically catalyzes the desaturation of L-Ile to the 3,4-dehydroisoleucine intermediate [5]. By providing this desaturated intermediate, PolE increases the flux through the PolF-catalyzed reaction, making the overall biosynthesis of PA more specific and efficient [5]. This explains the genetic evidence showing that a polE knockout strain still produces polyoxin, but at a significantly reduced titer [5].
Experimental Workflow and Methodologies
This section provides detailed protocols for key experiments used to characterize PolF, from gene to mechanism. The overall workflow is summarized in the diagram below.
Diagram 2: Integrated experimental workflow for characterizing PolF function, encompassing genetics, biochemistry, and structural biology.
Genetic Inactivation and Metabolite Analysis
- Method: In-frame deletion of polE and polF genes in Streptomyces cacaoi [5].
- Procedure: The target gene is replaced with a selectable marker via homologous recombination. Mutants are selected and verified by genomic analysis. The wild-type and mutant strains are cultured under polyoxin-producing conditions [5].
- Analysis: Fermentation broths are analyzed by Liquid Chromatography-Mass Spectrometry (LC-MS). The presence or absence of polyoxin A is determined by comparing chromatographic retention times and mass spectra with authentic standards [5].
In Vitro Enzyme Activity Assay
- Enzyme Preparation: Apo-PolF is purified from E. coli. The enzyme is reconstituted by incubation with a 3-fold molar excess of Fe(II) under anaerobic conditions (e.g., in an anaerobic glovebox). Unbound iron is removed using a desalting column [5].
- Reaction Setup: Inside the anaerobic chamber, PolF is mixed with substrate (e.g., L-Ile, L-Val) in a suitable buffer. The reaction is initiated by transferring the mixture to an Oâ‚‚-saturated buffer outside the chamber [5].
- Detection and Quantification:
- Derivatization: Reaction products can be derivatized with dansyl chloride (DnsCl) to improve detection [5].
- LC-MS Analysis: Products are separated by reverse-phase LC and detected by MS. Identification is based on mass loss (-4 Da for azetidine products) and comparison to authentic standards. Quantification can be achieved via calibration curves [5].
- Single-Turnover Experiments: Conducted with a stoichiometric amount of Fe(II) (2 eq. per PolF) and no external reductant to trap and observe reaction intermediates [5].
Structural and Mechanistic Probes
- Crystallography: X-ray crystallography of PolF, particularly in complex with substrates like L-Ile, provides atomic-level insights into the active site architecture and substrate binding mode, informing the mechanism of C–N bond formation [5]. Advanced methods like time-resolved crystallography and automated multiconformer model building (e.g., with tools like qFit) can further reveal dynamic changes and conformational heterogeneity during catalysis [14] [15].
- Spectroscopic Characterization: Techniques such as Mössbauer and electron paramagnetic resonance (EPR) spectroscopy are used to characterize the diiron cluster and identify reactive oxygen intermediates (e.g., the μ-peroxo-Fe(III)₂ species) during the catalytic cycle [5].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Studying PolF and Related Iron-Dependent Enzymes
| Reagent / Material | Function in Research | Specific Example / Note |
|---|---|---|
| Apo-PolF Enzyme | Catalytic core for in vitro functional studies | Recombinantly expressed and purified from E. coli [5] |
| Fe(II) Salts (e.g., Fe(NHâ‚„)â‚‚(SOâ‚„)â‚‚) | Reconstitution of the active diiron cofactor | Must be handled under anaerobic conditions to prevent oxidation [5] |
| External Reductants (Ascorbate, DTT) | Maintains diiron center in reduced state for multiple catalytic turnovers | Essential for sustained enzyme activity in assays [5] |
| Deuterated Solvents & NMR Standards | Structural elucidation of novel products and intermediates | Used for NMR characterization of PA, MAA, and other intermediates [5] |
| Derivatization Agents (e.g., Dansyl Chloride) | Enhances detection sensitivity for LC-MS analysis | Facilitates identification and quantification of amino acid-based products [5] |
| Synchrotron Radiation | High-intensity X-ray source for protein crystallography | Enables high-resolution structure determination of PolF and complexes [16] |
| Automated Model Building Software (e.g., qFit) | Models conformational heterogeneity from high-resolution crystallographic or cryo-EM data | Identifies alternative protein conformations relevant to catalysis [15] |
| XL413 hydrochloride | XL413 hydrochloride, CAS:1169562-71-3, MF:C14H13Cl2N3O2, MW:326.2 g/mol | Chemical Reagent |
| SB-277011 dihydrochloride | SB-277011 dihydrochloride, MF:C28H32Cl2N4O, MW:511.5 g/mol | Chemical Reagent |
The identification and characterization of PolF as a non-haem iron-dependent HDO enzyme has unveiled a novel and remarkable biocatalytic strategy for the formation of the strained azetidine ring. Unlike previously known mechanisms, PolF utilizes a μ-peroxo-Fe(III)₂ intermediate to drive a radical-mediated process that directly transforms simple L-amino acids into azetidine carboxylates. Its catalytic prowess, which includes desaturation, hydroxylation, and C–N bond formation activities on a single platform, coupled with the auxiliary desaturation function of PolE, provides a more efficient and potentially generalizable route to these valuable structures. The insights gained from the genetic, enzymological, and structural studies of PolF not only solve a long-standing biosynthetic puzzle but also significantly expand the known catalytic repertoire of the HDO superfamily. For researchers in drug development and synthetic biology, PolF represents a promising biocatalytic tool for the sustainable production of azetidine-containing building blocks, opening new avenues for the creation of pharmaceuticals and fine chemicals.
Mechanistic Insights and Practical Applications of Non-Haem Iron Enzymes
This technical guide outlines integrated genetic and enzymological workflows for characterizing biosynthetic pathways, using the recent elucidation of azetidine amino acid biosynthesis by non-haem iron-dependent enzymes as a foundational case study. We provide detailed methodologies for gene knockout, enzyme production, functional characterization, and structural analysis tailored to researchers investigating specialized metabolic pathways. The protocols emphasize practical considerations for handling oxygen-sensitive metalloenzymes and detecting transient reaction intermediates, with specific application to the PolF and PolE enzymes responsible for azetidine ring formation in polyoxin antifungal compounds.
The biosynthesis of azetidine-containing amino acids represents a significant biochemical challenge due to the high ring strain (25.4 kcal molâ»Â¹) characteristic of four-membered heterocycles [5]. Recent breakthroughs in understanding polyoximic acid biosynthesis in Streptomyces cacaoi have revealed novel non-haem iron-dependent enzymes capable of catalyzing this energetically demanding transformation [5] [6] [7]. This guide frames core genetic and enzymological techniques within this research context, providing standardized workflows for pathway validation and enzyme characterization that can be applied to similar biosynthetic systems.
The azetidine ring is a crucial structural element in many bioactive compounds and drugs, yet its biosynthetic origins have remained largely enigmatic until recent studies identified PolF as a haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily member capable of transforming L-isoleucine and L-valine into their azetidine derivatives via a 3,4-desaturated intermediate [5]. The accompanying PolE enzyme, a DUF6421 family member, functions as an Fe and pterin-dependent oxidase that increases flux through the pathway by catalyzing desaturation of L-Ile [5].
Genetic Workflow: Gene Knockout and Functional Analysis
Gene Knockout Strategies
Gene knockout techniques enable researchers to determine gene function by observing phenotypic consequences of specific gene inactivation [17]. In the context of azetidine biosynthesis, knockout experiments established the essential role of polF in polyoximic acid formation [5].
2.1.1 CRISPR-Cas9 Mediated Knockout CRISPR-Cas9 has revolutionized gene knockout efficiency across diverse organisms [18]. The system utilizes a guide RNA (gRNA) to direct the Cas9 nuclease to a specific genomic locus, creating a double-strand break (DSB) that is repaired predominantly through error-prone non-homologous end joining (NHEJ), resulting in frameshift mutations and gene inactivation [18] [19].
Critical Design Considerations:
- Target Selection: For complete knockout, target early exons common to all transcript variants [19]
- Off-target Assessment: Use tools like CRISPOR or CRISPRdirect to minimize off-target effects [19]
- Critical Exon Strategy: Delete exons meeting these criteria: present in all splicing variants, total nucleotide count not a multiple of 3, >50 bp from termination codon, and disruption destroys ≥50% of coding sequence [19]
2.1.2 Homologous Recombination Traditional homologous recombination-based methods remain valuable for specific applications, particularly when large deletions or precise modifications are required [17]. This approach involves creating a DNA construct with long homology arms flanking a drug selection marker, which replaces the target gene via the cell's native repair mechanisms [17].
Experimental Protocol: Microbial Gene Knockout
The following protocol was applied successfully in Streptomyces cacaoi to elucidate azetidine biosynthesis genes [5]:
Step 1: Target Identification and gRNA Design
- Identify target gene sequence within biosynthetic gene cluster
- Design gRNAs targeting critical exons or functional domains
- For polF and polE, target sequences within DUF6202 and DUF6421 domains respectively
Step 2: Vector Construction
- Clone selected gRNAs into appropriate Cas9-expression vector
- Include selection markers (antibiotic resistance) for stable transformants
- Incorporate inducible systems for essential gene analysis
Step 3: Transformation and Selection
- Introduce construct into host via electroporation or conjugation
- Plate on selective media and incubate until colonies form
- Screen colonies by PCR for desired genomic modifications
Step 4: Phenotypic Analysis
- Culture knockout mutants under polyoxin-producing conditions [5]
- Analyze metabolites via LC-MS comparing knockout to wild-type
- For polF knockout: complete loss of polyoxin A production (<1% of wild-type) [5]
- For polE knockout: significantly reduced but detectable polyoxin A (~10% of wild-type) [5]
Step 5: Genotype Validation
- Sequence target locus to confirm intended mutation
- Verify absence of off-target modifications through whole-genome sequencing
- Assess expression levels of flanking genes to rule out polar effects
Data Interpretation and Validation
Knockout results must be interpreted cautiously, as compensatory mechanisms may mask phenotypic effects [17]. For essential genes, conditional knockouts using Cre-loxP or similar systems enable temporal control of gene expression [17]. In the azetidine pathway, the differential phenotypes of polE versus polF knockouts revealed their distinct functional contributions, with PolF being essential while PolE serves an auxiliary role [5].
Enzymological Workflow: Recombinant Enzyme Production and Characterization
Enzyme Production and Purification
Functional characterization begins with recombinant production of catalytically active enzyme [5]:
Expression in E. coli:
- Clone gene into appropriate expression vector (e.g., pET series)
- Transform into expression strain (e.g., BL21(DE3))
- Induce expression with IPTG at optimal temperature (often 16-18°C for metalloenzymes)
- Harvest cells by centrifugation and lyse via sonication or French press
Purification of Metalloenzymes:
- Purify via affinity chromatography (His-tag, GST-tag, etc.)
- For iron-dependent enzymes like PolF: perform under anaerobic conditions when necessary
- Remove unbound metal via desalting chromatography
- Reconstitute apo-enzyme with excess Fe(II) (3 eq.) under anaerobic conditions
- Remove excess metal and verify incorporation (PolF contained 1.5 eq. Fe(II) after reconstitution) [5]
Enzyme Activity Assays
Standard Activity Assay for PolF-like Enzymes [5]:
- Prepare assay mixture anaerobically in glove box
- Include: enzyme (PolF), substrate (L-Ile or L-Val), excess Fe(II), buffer
- Initiate reaction by adding Oâ‚‚-saturated buffer
- Terminate reaction at appropriate timepoints
- Derivatize products with dansyl chloride (DnsCl) for detection
- Analyze via LC-MS
Key Controls:
- No enzyme (background reaction)
- No Fe(II) (metal dependence)
- No Oâ‚‚ (oxygen dependence)
- Heat-inactivated enzyme (enzyme dependence)
- Alternative metals (specificity)
Table 1: Substrate Specificity of PolF
| Substrate | Major Product | Relative Activity | Notes |
|---|---|---|---|
| L-isoleucine | Polyoximic acid (PA) | 100% | Natural substrate |
| L-valine | 3-methylene-azetidine-2-carboxylic acid (MAA) | ~80% | Forms azetidine ring |
| L-leucine | Hydroxylation products | <5% | Minor desaturation |
| L-methionine | Hydroxylation products | <5% | Minor desaturation |
| L-allo-Ile | Azetidine products | <10% | Stereochemistry important |
| Other proteogenic amino acids | No detectable products | - | High specificity |
Kinetic Characterization
Establish kinetic parameters using optimized assay conditions [20]:
Initial Velocity Measurements:
- Vary substrate concentration while maintaining saturating other components
- Determine initial rates at each substrate concentration
- Plot data and fit to appropriate model (Michaelis-Menten, etc.)
Advanced Kinetic Analysis:
- Pre-steady-state kinetics for mechanistic studies
- Single-turnover experiments to isolate specific steps
- For PolF: single-turnover revealed 3,4-dh-Val as major intermediate (0.23 minâ»Â¹) followed by Azi, 4-OH-Val and 3-OH-Val (0.15 minâ»Â¹, 0.14 minâ»Â¹ and 0.036 minâ»Â¹) [5]
Reductant Dependence: Assess requirement for external reductants (ascorbate, DTT) which are often necessary for multiple turnovers in HDO enzymes [5].
Structural and Mechanistic Characterization
Spectroscopic Methods
Multiple spectroscopic techniques provide insight into metalloenzyme active sites:
UV-Visible Spectroscopy [20]:
- Monitor reaction progress and intermediate formation
- Identify charge-transfer bands characteristic of metal centers
Advanced Spectroscopic Techniques:
- Mössbauer spectroscopy for iron oxidation and coordination states
- EPR for paramagnetic intermediates
- For PolF: identification of μ-peroxo-Fe(III)₂ intermediate responsible for C–H cleavage [5]
Structural Biology Approaches
X-ray Crystallography [20]:
- Grow high-quality crystals of native and substrate-bound forms
- Collect diffraction data at synchrotron sources
- Solve structure by molecular replacement or experimental phasing
- For PolF: structures revealed active site architecture and substrate positioning [5]
Alternative Structural Methods:
- Cryo-EM for large complexes or difficult-to-crystallize proteins
- NMR for solution dynamics and conformational changes [20]
Reaction Intermediate Analysis
Trapping and Identification:
- Use single-turnover conditions with limited Oâ‚‚
- Quench reactions at various timepoints
- Analyze intermediates via LC-MS and NMR
- For PolF: identified 3,4-dehydrovaline (3,4-dh-Val) as key intermediate quantitatively converted to MAA [5]
Mechanistic Probes:
- Isotope labeling (²H, ¹â¸O) to track atom fate
- Substrate analogs to test specificity
- Radical traps to identify radical intermediates
Biosynthesis of Azetidine Amino Acids: A Case Study
The biosynthesis of polyoximic acid in Streptomyces cacaoi provides a comprehensive example integrating these genetic and enzymological approaches [5].
Early isotope-labelling experiments indicated L-isoleucine as the polyoximic acid precursor, suggesting a novel biosynthetic mechanism distinct from SAM-dependent or α-ketoglutarate-dependent routes [5]. Gene cluster analysis identified polE and polF as candidate biosynthetic genes, though their specific functions were unknown.
Functional Characterization of PolF
Genetic knockout established PolF as essential for polyoximic acid biosynthesis [5]. In vitro reconstitution demonstrated that PolF alone could convert L-Ile to polyoximic acid, with absolute requirement for Fe(II) and Oâ‚‚ [5]. Substrate specificity studies revealed that PolF accepts L-Ile and L-Val as primary substrates, generating azetidine products, while other amino acids yield only minor hydroxylation products [5].
Mechanistic studies identified PolF as an HDO superfamily member that utilizes a μ-peroxo-Fe(III)₂ intermediate for unactivated C–H bond cleavage [5]. The subsequent C–N bond formation proceeds through a radical mechanism, with 3,4-desaturated intermediates serving as precursors to azetidine ring formation.
Role of PolE
While PolF alone can catalyze the complete transformation, PolE enhances pathway efficiency by catalyzing Fe and pterin-dependent desaturation of L-Ile, increasing flux through the pathway [5]. This auxiliary function explains the reduced but detectable polyoxin production in polE knockout strains.
Integrated Reaction Mechanism
The current mechanistic model for azetidine formation includes:
- Initial C–H activation by μ-peroxo-Fe(III)₂ species
- Formation of 3,4-desaturated intermediate
- Radical-mediated C–N bond formation
- Azetidine ring closure with concomitant reduction
Diagram 1: Azetidine Biosynthesis Pathway
Essential Research Reagents and Solutions
Table 2: Key Research Reagents for Azetidine Biosynthesis Studies
| Reagent/Solution | Function/Application | Specific Examples | Technical Notes |
|---|---|---|---|
| Genetic Engineering | |||
| CRISPR-Cas9 system | Targeted gene knockout | Streptococcus pyogenes Cas9 | Most widely used system [19] |
| Guide RNA (gRNA) | Targets Cas9 to specific locus | CRISPOR-designed sequences | Minimize off-target effects [19] |
| Homologous recombination vectors | Traditional gene replacement | pKO vectors with selection markers | Lower efficiency than CRISPR [17] |
| Enzyme Characterization | |||
| Affinity chromatography resins | Protein purification | Ni-NTA (His-tag), Glutathione (GST-tag) | Maintain anaerobic conditions for metalloenzymes |
| Iron supplements | Metalloenzyme reconstitution | Fe(II) salts (e.g., FeSOâ‚„) | Add anaerobically to prevent oxidation [5] |
| Anaerobic chamber | Oxygen-sensitive procedures | PolF assay setup | Essential for Fe(II)-dependent enzymes [5] |
| Analytical Tools | |||
| LC-MS systems | Metabolic and enzyme analysis | Q-TOF, Orbitrap instruments | Detect -4 Da mass change in azetidine formation [5] |
| NMR spectroscopy | Structural characterization of products | ¹H, ¹³C NMR | Confirm azetidine ring structure [5] |
| X-ray crystallography | Enzyme structure determination | Synchrotron sources | Reveal active site architecture [5] [20] |
| Specialized Reagents | |||
| Dansyl chloride (DnsCl) | Product derivatization for detection | PolF assay derivatization | Enhances LC-MS sensitivity [5] |
| Phosphoenolpyruvate | PEP-dependent reactions | Pyruvyltransferase assays | Substrate for pyruvylation [21] |
| UDP-sugar donors | Glycosyltransferase assays | SCWP biosynthesis studies | Lipid-linked repeat formation [21] |
The integration of genetic knockout strategies with detailed enzymological characterization provides a powerful framework for elucidating biosynthetic pathways, as demonstrated by the recent breakthroughs in understanding azetidine amino acid biosynthesis. The case study of PolF and PolE highlights how coordinated application of these techniques can unravel novel enzymatic mechanisms, even for chemically challenging transformations like four-membered ring formation.
These workflows establish a standardized approach applicable to diverse biosynthetic systems, particularly those involving metalloenzymes and specialized metabolism. The continued refinement of gene editing technologies coupled with advanced structural and mechanistic enzymology promises to accelerate the discovery and characterization of novel enzymatic functions with potential applications in drug development and biocatalysis.
The biosynthesis of azetidine, a strained four-membered nitrogen-containing heterocycle found in numerous bioactive compounds and drugs, has long posed a significant challenge to mechanistic understanding. Due to its high ring strain, enzymatic formation of azetidine represents a remarkable chemical transformation that has remained enigmatic [5]. Within the context of azetidine amino acid biosynthesis by non-haem iron-dependent enzymes, structural biology has played a pivotal role in elucidating the active site architecture and catalytic mechanisms responsible for these energetically challenging ring formations.
Recent research on the polyoxin antifungal pathway has revealed unprecedented enzymatic machinery capable of constructing these strained rings. The key enzyme PolF, a member of the haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily, has been shown to transform linear amino acids into azetidine derivatives via a radical-based mechanism [5] [6]. This discovery, facilitated by crystal structure determination of PolF in complex with its substrate, provides crucial insights into how enzymes overcome substantial kinetic and thermodynamic barriers to form constrained cyclic systems.
Structural Revelations from the PolF Crystal Structure
The X-ray crystal structure of Streptomyces cacaoi PolF in complex with iron and L-isoleucine (PDB ID: 9PRR) has provided an unprecedented view into the active site architecture responsible for azetidine formation [22]. Solved at 2.06 Ã… resolution, this structure reveals critical features that enable the remarkable cyclization reaction.
Active Site Architecture and Metal Coordination
The PolF active site features a dimetal center coordinated by histidine and carboxylate residues, characteristic of HDO superfamily enzymes [5]. The two iron atoms are positioned to activate molecular oxygen and generate the reactive species responsible for hydrogen atom abstraction. The L-isoleucine substrate binds in close proximity to this diiron center, positioned optimally for the initial C-H bond cleavage at the C3 position [5] [22].
Table 1: Key Structural Parameters of PolF Active Site (PDB 9PRR)
| Parameter | Description | Significance |
|---|---|---|
| Resolution | 2.06 Ã… | Allows precise positioning of substrate and metal ions |
| Fe-Fe Distance | ~3.5-4.0 Å | Optimal for μ-peroxo-Fe(III)₂ intermediate formation |
| Substrate Positioning | L-Ile bound near diiron center | Enables direct H-atom abstraction from C3 position |
| Metal Coordination | Histidine and carboxylate ligands | Typical for non-haem diiron enzymes |
| Access Channel | Hydrophobic pocket | Accommodates medium-chain aliphatic amino acids |
The structural data reveals how the enzyme orients the substrate through specific interactions with active site residues, particularly those recognizing the amino and carboxyl groups of the isoleucine substrate. This precise positioning is essential for the regio- and stereoselectivity observed in the azetidine formation [22].
Substrate Recognition and Specificity
The PolF structure in complex with L-isoleucine provides insights into the enzyme's substrate specificity. The active site accommodates medium-size aliphatic amino acids, with the highest activity observed for L-isoleucine and L-valine [5]. The β-methyl group of these substrates appears critical for azetidine formation, as L-methionine and L-leucine yield primarily hydroxylation products with minimal cyclization [5].
The structural constraints explain the observed stereochemical preferences, though the enzyme exhibits some flexibility toward isoleucine stereoisomers, with l-allo-Ile, d-Ile, and d-allo-Ile all yielding detectable azetidine products [5].
Catalytic Mechanism Revealed by Structural Data
The PolF crystal structure has enabled a detailed proposal for the azetidine ring formation mechanism, highlighting the role of the diiron center in facilitating this challenging transformation.
The Oxidative Initiation Step
Structural and spectroscopic evidence indicates that PolF utilizes a μ-peroxo-Fe(III)â‚‚ intermediate to initiate catalysis [5]. This species is directly responsible for the cleavage of the unactivated C3-H bond, which has a bond dissociation energy of up to 101 kcal molâ»Â¹ [5]. The crystal structure shows the proximity of this carbon center to the diiron cluster, supporting the feasibility of this hydrogen atom abstraction step.
Radical-Mediated C-N Bond Formation
Following the initial H-atom abstraction, the catalytic mechanism proceeds through a radical-based pathway for C-N bond formation. The structure reveals how the enzyme positions the developing carbon radical adjacent to the nitrogen atom, enabling the cyclization reaction to proceed. This process likely involves:
- Formation of a carbon-centered radical at C3 after hydrogen abstraction
- Intramolecular radical attack on the nitrogen atom
- Ring closure to form the azetidine structure
- Further oxidation to generate the final product [5]
The structural data supports this mechanism by demonstrating the spatial relationship between the C3 carbon and nitrogen atom in the bound substrate conformation.
Figure 1: Catalytic Mechanism of PolF Enzyme. The diagram illustrates the proposed reaction pathway for azetidine formation, from substrate binding through the key radical intermediate to cyclization.
Experimental Methods and Protocols
The elucidation of PolF's structure and mechanism employed a comprehensive suite of biochemical, structural, and spectroscopic techniques.
Protein Expression and Crystallization
Researchers expressed PolF in Escherichia coli BL21(DE3) and purified it using standard chromatographic techniques [22]. The enzyme was initially isolated in predominantly apo form, requiring reconstitution with iron under anaerobic conditions for functional studies [5]. Crystallization was achieved using the hanging-drop vapor-diffusion method, with glycerol serving as a cryoprotectant for data collection at cryogenic temperatures [22].
Table 2: Key Research Reagents and Experimental Solutions
| Reagent/Solution | Function/Application | Experimental Role |
|---|---|---|
| Apo-PolF | Iron-free enzyme preparation | Serves as starting material for metal reconstitution studies |
| Fe(II) sulfate | Metal cofactor source | Reconstitutes active diiron center in apo-PolF |
| L-Isoleucine | Native substrate | Primary substrate for azetidine formation assays |
| DnsCl (Dansyl chloride) | Derivatization agent | Enables LC-MS detection and quantification of reaction products |
| Oâ‚‚-saturated buffer | Oxidant source | Initiates catalytic turnover in anaerobic assays |
| Ascorbate/DTT | Reductant | External electron donor for multiple turnover conditions |
Structural Determination and Analysis
X-ray diffraction data were collected at synchrotron sources and processed using XDS and Aimless software packages [22]. The structure was solved using molecular replacement with PHENIX software, and iterative model building and refinement were performed using Coot and PHENIX [22]. The quality of the final model was validated using MolProbity and PDB validation tools.
Functional Characterization assays
Enzyme activity assays were conducted under both multiple turnover and single turnover conditions [5]. For standard assays, apo-PolF was reconstituted with excess Fe(II) under anaerobic conditions, then reactions were initiated by adding Oâ‚‚-saturated buffer. Products were derivatized with dansyl chloride and analyzed by LC-MS [5]. Single turnover experiments used stoichiometric Fe(II) without external reductants to isolate intermediate species.
Figure 2: Experimental Workflow for PolF Characterization. The diagram outlines the integrated approach combining genetics, biochemistry, and structural biology to elucidate PolF function.
Complementary Enzymatic Machinery: The Role of PolE
Structural and functional studies have also characterized PolE, a member of the DUF6421 family that assists PolF in the azetidine biosynthetic pathway. PolE is an iron and pterin-dependent oxidase that catalyzes the desaturation of L-isoleucine, enhancing the flux through the azetidine formation pathway by increasing the concentration of the 3,4-desaturated intermediate [5] [11].
While the crystal structure of PolE is not yet publicly available, biochemical studies indicate that it functions synergistically with PolF by providing a more efficient route to the desaturated intermediate that serves as a precursor for azetidine formation [5]. This division of labor between two non-haem iron enzymes represents an elegant biosynthetic strategy for managing the challenging reaction coordinates of strained ring formation.
Implications for Drug Discovery and Development
The structural insights from PolF have significant implications for pharmaceutical applications. Azetidine rings are valuable scaffolds in drug design, featured in antibiotics, antiviral agents, and cancer therapeutics [23]. The traditional chemical synthesis of these strained rings often requires harsh solvents and toxic chemicals, making the enzymatic route an attractive alternative for sustainable pharmaceutical manufacturing [23].
The discovery of PolF's ability to produce both azetidine (four-membered) and aziridine (three-membered) rings from inexpensive precursor compounds presents unprecedented opportunities for biocatalytic applications [23]. With chemical precursors for existing azetidine synthesis methods costing approximately $1,390 per gram, the PolF system offers a potentially more economical and environmentally friendly alternative [23].
The crystal structure of PolF has provided unprecedented insights into the active site architecture and catalytic mechanism of azetidine amino acid biosynthesis. By revealing the dimetal center responsible for activating molecular oxygen and the substrate-binding pocket that positions L-isoleucine for the challenging cyclization reaction, structural biology has illuminated how enzymes overcome substantial kinetic and thermodynamic barriers to form strained heterocyclic systems.
These findings not only solve a long-standing puzzle in natural product biosynthesis but also open new avenues for biocatalytic applications in pharmaceutical synthesis. The structural blueprint of PolF's active site may guide future engineering efforts to develop tailored catalysts for producing diverse azetidine-containing compounds with therapeutic potential. As the field advances, the integration of structural biology with mechanistic enzymology will continue to reveal nature's sophisticated solutions to challenging chemical transformations.
The μ-peroxo-Fe(III)â‚‚ intermediate is a pivotal species in the catalytic cycles of non-haem diiron enzymes, serving as the primary agent for the reductive activation of dioxygen (Oâ‚‚) to perform challenging oxidative transformations in biological systems [24]. This intermediate is characterized by a peroxo bridge (O₂²â») coordinating two ferric (Fe(III)) ions within a single enzyme active site. Its formation enables enzymes to cleach exceptionally strong C–H bonds, a fundamental step in the biosynthesis of complex natural products [25] [5]. Within the context of azetidine amino acid biosynthesis, this intermediate is instrumental in initiating a radical-based reaction cascade that forges the strained four-membered ring, a structure of significant pharmaceutical interest [5] [7]. This whitepaper provides an in-depth technical examination of the μ-peroxo-Fe(III)â‚‚ intermediate, detailing its formation, structural and spectroscopic properties, and its direct role in C–H bond activation, with a specific focus on the novel biosynthetic pathway for polyoximic acid.
Structural and Spectroscopic Properties of the μ-peroxo-Fe(III)₂ Intermediate
The μ-peroxo-Fe(III)₂ intermediate is not a single monolithic entity; its stability, geometry, and reactivity are finely tuned by the enzyme's active site architecture. A detailed understanding of its properties is essential for identifying and characterizing this species in enzymatic studies.
Core Structure and Geometric Isomerism
The core unit of this intermediate is the {FeIII(μ-O)(μ-1,2-O2)FeIII} structure, which features both a single μ-oxo bridge and a μ-1,2-peroxo bridge [26]. In the μ-1,2 (or "side-on") binding mode, the peroxo ligand bridges the two iron atoms, with each oxygen atom bound to a different iron ion. This geometry is distinct from the μ-1,1 (or "end-on") configuration. The O–O bond length in synthetic μ-1,2-peroxo model complexes has been measured between 1.396 and 1.432 Ã…, which is characteristic of a peroxo (O₂²â») ligand and is notably longer than the O–O bond in superoxo (Oâ‚‚â») or free dioxygen [26].
Spectroscopic Signatures
Key spectroscopic features allow researchers to unambiguously identify the μ-peroxo-Fe(III)₂ intermediate and distinguish it from other high-valent iron-oxo species.
- UV-Vis-NIR Spectroscopy: These intermediates typically exhibit intense absorption bands in the visible to near-infrared region due to ligand-to-metal charge transfer (LMCT). A prominent band between 14,000 and 15,000 cmâ»Â¹ is a characteristic feature assigned as the μ-1,2-peroxo→FeIII LMCT transition [25] [26] [24]. For instance, the intermediate in Pseudomonas fluorescens UndA has a λmax at ~550 nm (~18,200 cmâ»Â¹) [25], while a synthetic model complex showed LMCT bands at 15,400 cmâ»Â¹ and 19,300 cmâ»Â¹ [26].
- Mössbauer Spectroscopy: Mössbauer spectra provide critical information on the oxidation and spin state of the iron centers. The μ-peroxo-Fe(III)₂ intermediate exhibits isomer shifts (δ) characteristic of high-spin Fe(III) but often at the higher end of the typical range (δ ~ 0.48–0.68 mm/s), reflecting the covalency of the Fe–Operoxo bond [25] [26]. The spectra typically show quadrupole doublets, and the intermediate is usually diamagnetic or anti-ferromagnetically coupled, resulting in an integer-spin or S=0 ground state [25].
- Resonance Raman (rR) Spectroscopy: Excitation into the peroxo-to-Fe(III) LMCT band enhances vibrations associated with the peroxo ligand. The O–O stretching vibration (νO–O) is a key indicator, typically found in the range of ~850–900 cmâ»Â¹ [24]. Isotopic labeling with ¹â¸Oâ‚‚ confirms the assignment of this vibration.
Table 1: Key Spectroscopic Parameters of Characterized μ-peroxo-Fe(III)₂ Intermediates
| Enzyme/Model System | UV-Vis LMCT Band (cmâ»Â¹) | Mössbauer δ (mm/s) | νO–O (cmâ»Â¹) | Reference |
|---|---|---|---|---|
| P. fluorescens UndA | ~18,200 | 0.59, 0.56 | Not Reported | [25] |
| Synthetic {FeIII(μ-O)(μ-1,2-O2)FeIII} | 15,400, 19,300 | 0.53 | ~850-900 (expected) | [26] |
| Soluble Methane Monooxygenase | ~14,000-15,000 | ~0.50-0.68 | ~850-900 | [24] |
The Catalytic Cycle in C-H Activation: A Step-by-Step Analysis
The catalytic cycle of non-haem diiron enzymes employing the μ-peroxo-Fe(III)₂ intermediate follows a conserved sequence, from O₂ binding to substrate oxidation. The following diagram illustrates the core mechanistic pathway.
Step 1: Formation of the Diferrous Reactant Complex
The cycle begins with the enzyme in its diferrous state (Feâ‚‚(II/II)). For some enzymes, such as UndA, substrate binding is a prerequisite for the efficient uptake of two Fe(II) ions and the proper assembly of the cofactor [25] [5]. The substrate binds in proximity to the diiron site, priming the enzyme for Oâ‚‚ activation.
Step 2: Oâ‚‚ Activation and Peroxo Intermediate Formation
Dioxygen (O₂) binds to the Fe₂(II/II) cluster, leading to a two-electron reduction of O₂ and the formation of the μ-peroxo-Fe₂(III/III) intermediate. This step is rapid and, in cases like the UndA enzyme, is triggered by substrate binding, which likely induces a conformational change that optimizes the cluster for O₂ binding and activation [25]. The kinetics of this formation are O₂-concentration dependent [25].
Step 3 & 4: Substrate Positioning and C-H Bond Cleavage
The μ-peroxo-Feâ‚‚(III/III) intermediate is directly responsible for the cleavage of unactivated C–H bonds. In the PolF enzyme, this intermediate abstracts a hydrogen atom from the C3 position of L-isoleucine or a 3,4-desaturated intermediate [5]. This Hydrogen Atom Transfer (HAT) step is remarkable because it targets C–H bonds with high homolytic bond dissociation energies (BDEs), estimated to be up to 101 kcal molâ»Â¹ [5]. The ability to cleave such strong bonds underscores the potent reactivity of this intermediate, which is enabled by the electronic structure of the Fe–O–O–Fe unit.
Step 5: Product Formation and Cluster Reduction
Following HAT, the resulting carbon-centered radical undergoes further transformations. In PolF, this leads to intramolecular C–N bond formation to construct the azetidine ring, presumably through radical recombination mechanisms [5]. This step completes the oxidative decarboxylation or cyclization reaction, leaving the diiron cluster in its oxidized Fe₂(III/III) state. For multiple turnovers, an external reductant such as ascorbate is required to return the cluster to the diferrous Fe₂(II/II) state [5].
Experimental Protocols for Intermediate Characterization
The study of transient species like the μ-peroxo-Fe₂(III/III) intermediate requires specialized rapid-mixing techniques and multi-faceted spectroscopic approaches.
Anaerobic Protein Reconstitution and Sample Preparation
- Buffer Preparation: Prepare all buffers in an anaerobic glove box (Oâ‚‚ < 1 ppm) or thoroughly degas them by bubbling with an inert gas (Ar or Nâ‚‚). Use buffers such as 50 mM HEPES, pH 7.5.
- Apo-Enzyme Incubation: Incubate the purified apo-enzyme (e.g., PolF or UndA) with a 2-3 molar equivalents of Fe(II) (e.g., Fe(NHâ‚„)â‚‚(SOâ‚„)â‚‚) under anaerobic conditions [5].
- Removal of Excess Iron: Pass the reconstituted enzyme through a desalting column (e.g., PD-10) equilibrated with anaerobic buffer to remove unbound Fe(II). Determine the iron-to-protein stoichiometry by colorimetric assay or inductively coupled plasma mass spectrometry (ICP-MS). A ratio of ~1.5-2.0 is typical for a diiron cluster [25] [5].
- Substrate Addition: For substrate-triggered reactions, add the substrate (e.g., dodecanoic acid for UndA, L-Ile for PolF) to the anaerobic enzyme solution.
Rapid Kinetics and Freeze-Quench Mössbauer Spectroscopy
This protocol is used to trap and characterize short-lived intermediates.
- Rapid Mixing: Use a stopped-flow or rapid-freeze-quench apparatus. Load one syringe with the anaerobic, substrate-bound diiron(II) enzyme complex. Load the second syringe with Oâ‚‚-saturated buffer.
- Reaction Initiation and Quenching: Rapidly mix the solutions at the desired temperature (e.g., 4°C). For freeze-quench, the reaction mixture is extruded directly into a cryogenic liquid (e.g., liquid isopentane at ~ -130°C) at specific time points (e.g., 0.01 s to several seconds) [25].
- Mössbauer Data Collection: Pack the frozen quenched samples into a Mössbauer spectrometer cup. Collect spectra over a range of temperatures and applied magnetic fields. The spectra of the intermediate (e.g., at 0.01 s) will show new quadrupole doublets with isomer shifts characteristic of high-spin Fe(III), which can be simulated to determine δ and ΔE_Q [25].
UV-Vis Spectroscopy for Intermediate Detection
- Stopped-Flow UV-Vis Setup: Use a diode-array stopped-flow spectrophotometer housed inside an anaerobic glove box or equipped with anaerobic capabilities.
- Data Acquisition: Mix the anaerobic diiron(II) enzyme (with or without substrate) with Oâ‚‚-saturated buffer. Record full UV-Vis spectra in rapid succession (millisecond timescale).
- Data Analysis: Identify the formation and decay of transient species by their characteristic absorption features. For the μ-peroxo-Fe₂(III/III) intermediate, monitor the growth and decay of the ~550 nm (for UndA) or similar feature [25]. Plot the absorbance change over time to determine the observed rate constant (k_obs) for formation and decay.
The Scientist's Toolkit: Key Research Reagents and Materials
Successful investigation of μ-peroxo-Fe₂(III/III) intermediates requires a carefully selected set of reagents and analytical tools.
Table 2: Essential Reagents and Materials for Diiron Enzyme Studies
| Reagent/Material | Function and Specific Role | Example from Literature |
|---|---|---|
| Fe(II) Salts | Reconstitution of the diiron cofactor into the apo-enzyme. | Fe(NHâ‚„)â‚‚(SOâ‚„)â‚‚ [5] |
| Anaerobic Chamber | Provides an oxygen-free environment for protein reconstitution and sample preparation to prevent uncontrolled oxidation. | Used in all enzymatic studies of Oâ‚‚-sensitive intermediates [25] [5] |
| Stopped-Flow Spectrophotometer | Enables rapid mixing and real-time monitoring of transient intermediate formation and decay on millisecond timescales. | Used to detect the 550 nm transient in UndA [25] |
| Freeze-Quench Apparatus | Physically traps reactive intermediates at specific time points for analysis by non-time-resolved methods (e.g., Mössbauer, EPR). | Used to trap the μ-peroxo-Fe₂(III/III) intermediate in UndA for Mössbauer spectroscopy [25] |
| Ascorbic Acid / Dithiothreitol (DTT) | Acts as an external reductant to recycle the diiron cofactor from the Feâ‚‚(III/III) back to the Feâ‚‚(II/II) state for multiple enzyme turnovers. | Required for multiple turnovers of PolF [5] |
| âµâ·Fe-Enriched Iron | Isotopic enrichment for Mössbauer spectroscopy, significantly enhancing the signal-to-noise ratio. | Used for high-quality Mössbauer data collection [25] |
| (S,R,S)-AHPC-PEG4-N3 | (S,R,S)-AHPC-PEG4-N3, MF:C32H47N7O8S, MW:689.8 g/mol | Chemical Reagent |
| N-Nitrosoanatabine-d4 | N-Nitrosoanatabine-d4, CAS:1020719-69-0, MF:C10H11N3O, MW:193.24 g/mol | Chemical Reagent |
Case Study: Azetidine Ring Formation in Polyoximic Acid Biosynthesis
The biosynthesis of the azetidine-containing amino acid polyoximic acid (PA) in the polyoxin pathway provides a compelling case of the μ-peroxo-Fe₂(III/III) intermediate driving a complex multi-step transformation. The enzyme PolF, a member of the haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily, catalyzes the conversion of L-isoleucine (L-Ile) to PA [5].
Genetic and biochemical experiments confirm that PolF is essential for PA production [5]. In vitro assays with purified PolF, Fe(II), and O₂ successfully convert L-Ile to PA, with the reaction requiring an external reductant like ascorbate for multiple turnovers [5]. The proposed mechanism involves the μ-peroxo-Fe₂(III/III) intermediate of PolF directly cleaving a strong, unactivated C–H bond. This HAT event initiates a radical cascade that leads to desaturation, hydroxylation, or C–N bond formation, ultimately resulting in azetidine ring closure [5]. The ability of PolF to also convert L-valine to 3-methylene-azetidine-2-carboxylic acid (MAA) further demonstrates its capability for C–H activation and subsequent cyclization [5].
The μ-peroxo-Fe(III)₂ intermediate is a cornerstone of catalysis in a growing family of non-haem diiron enzymes. Its defined spectroscopic signatures and potent ability to activate strong C–H bonds make it a critical species in diverse biosynthetic pathways. The recent elucidation of its role in the enzymatic construction of strained azetidine rings by PolF not only solves a long-standing biosynthetic puzzle but also opens new avenues for bioinspired catalyst design [5] [7]. A deep understanding of its catalytic cycle, supported by advanced spectroscopic and kinetic techniques, provides researchers with the fundamental knowledge to explore novel enzymatic functions and harness these powerful biological catalysts for synthetic applications in chemistry and medicine.
The azetidine ring, a strained four-membered aza-cycle, is a crucial structural motif in numerous bioactive compounds and pharmaceuticals. Despite its significance, the biosynthetic pathways governing its formation have remained largely enigmatic, primarily due to the substantial ring strain (approximately 25.4 kcal molâ»Â¹) that makes its construction energetically challenging [5]. Within the broader thesis on the biosynthesis of azetidine-containing natural products, this guide delineates a novel enzymatic mechanism elucidated in the polyoxin antifungal pathway. Polyoxin A contains a distinctive azetidine amino acid known as polyoximic acid (PA), which was previously known to be derived from L-isoleucine (L-Ile) but whose transformation mechanism was undefined [5] [11]. Recent research has identified that two non-haem iron-dependent enzymes, PolE and PolF, are central to this transformation, operating via a fascinating desaturation and radical-mediated cyclization sequence [5] [27]. This pathway represents a significant departure from previously characterized mechanisms that rely on expensive precursors like S-adenosyl-L-methionine (SAM) or α-ketoglutarate (α-KG), thereby offering new, more efficient biocatalytic routes for azetidine synthesis [5].
Key Enzymes and Experimental Approaches
The Catalytic Players: PolF and PolE
The biosynthesis of polyoximic acid is orchestrated by two principal enzymes, each belonging to a unique family of non-haem iron-dependent enzymes and playing a distinct role.
PolF: The Central Azetidine-Forming Enzyme PolF is a member of the haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily. It is the core catalyst that alone is sufficient for the transformation of L-Ile and L-valine (L-Val) into their corresponding azetidine derivatives [5]. Genetic knockout experiments confirmed its indispensability, as a polF mutant failed to produce any measurable polyoxin A (<1% of wild-type) [5]. PolF is a diiron-dependent enzyme that activates molecular oxygen to perform chemically challenging C–H bond cleavages and subsequent C–N bond formation, likely via radical mechanisms [5] [27].
PolE: The Desaturation Assistant PolE, a member of the DUF6421 family, functions as an auxiliary enzyme. It is an Fe and pterin-dependent oxidase that specifically catalyses the desaturation of L-Ile to form a 3,4-dehydroisoleucine intermediate [5] [11]. While not absolutely essential for polyoxin production—a polE mutant still produces about 10% of the wild-type titre—it significantly increases the metabolic flux towards PA by providing a more readily cyclized substrate for PolF [5].
Essential Research Reagents and Methodologies
A detailed understanding of this biosynthetic pathway was achieved through a combination of genetic, enzymological, and structural experiments. The table below summarizes the key reagents and methodologies central to this research.
Table 1: Key Experimental Reagents and Methodologies for Studying Azetidine Biosynthesis
| Reagent/Method | Function/Description | Key Experimental Insight |
|---|---|---|
| Gene Knockout (in S. cacaoi) | In-frame deletion of polE or polF to assess their necessity in polyoxin A production [5]. | Confirmed PolF is essential; PolE enhances titre [5]. |
| In vitro Assay with Apo-PolF | Anaerobic reconstitution of purified apo-PolF with Fe(II), initiation of reaction with Oâ‚‚-saturated buffer [5]. | Demonstrated PolF's sufficiency to convert L-Ile to PA; confirmed Fe(II) and Oâ‚‚ dependence [5]. |
| Product Derivatization (DnsCl) | Derivatization of reaction products with dansyl chloride for LC–MS analysis [5]. | Enabled detection and identification of PA and other reaction intermediates [5]. |
| Single vs. Multiple Turnover Assays | Single turnover: limited Fe(II), no reductant. Multiple turnover: excess Fe(II) with reductant (ascorbate/DTT) [5]. | Revealed kinetic hierarchy of intermediates; confirmed need for external reductant in multiple turnovers [5]. |
| X-ray Crystallography | Determination of crystal structures of PolF, including in complex with L-Ile [5] [11]. | Revealed active site architecture and substrate binding mode, informing the mechanism of C–N bond formation [5]. |
| Spectroscopic Characterization | Analysis of the diiron cluster in PolF [5]. | Identified the μ-peroxo-Fe(III)₂ species as the key intermediate responsible for initial C–H bond cleavage [5]. |
The Catalytic Workflow: From Amino Acid to Azetidine
The transformation of a linear amino acid into a strained azetidine ring involves a precisely orchestrated sequence of steps. The following diagram maps the entire experimental and catalytic workflow, from enzyme preparation to azetidine ring formation.
Stepwise Mechanism of Azetidine Ring Formation
Initial Substrate Activation: The Role of Desaturation
The journey to the azetidine ring begins with the activation of the aliphatic amino acid substrate, L-Ile or L-Val. PolF can directly act on these substrates, but the pathway is enhanced by the prior action of PolE.
- PolE-Catalyzed Desaturation: PolE, an Fe and pterin-dependent oxidase, specifically catalyzes the removal of two hydrogen atoms from L-Ile to generate a 3,4-dehydroisoleucine intermediate (a -2 Da mass change) [5]. This desaturation step introduces a double bond between C3 and C4, priming the substrate for subsequent cyclization by reducing the energy barrier for the radical reactions catalyzed by PolF.
- Direct PolF Substrate Entry: In the absence of PolE, PolF can itself perform this desaturation. Under single-turnover conditions (with no external reductant), the 3,4-dehydro intermediate (e.g., 3,4-dh-Val from L-Val) is the major initial product observed [5]. This indicates that desaturation is the first committed step in the PolF catalytic cycle.
Table 2: Quantitative Analysis of PolF Substrate Specificity and Product Distribution with L-Val
| Parameter | L-Valine | L-Isoleucine | L-Leucine | L-Methionine |
|---|---|---|---|---|
| Primary Azetidine Product | 3-Methylene-azetidine-2-carboxylic acid (MAA) [5] | Polyoximic Acid (PA) [5] | Not formed [5] | Not formed [5] |
| Observed Intermediates | 3,4-dehydrovaline (3,4-dh-Val), 3-OH-Val, 4-OH-Val, Aziridine (Azi) [5] | Not Specified | Hydroxylation products (minor desaturation) [5] | Hydroxylation products (minor desaturation) [5] |
| Key Structural Requirement | β-methyl group is critical for azetidine formation [5] | β-methyl group is critical for azetidine formation [5] | N/A | N/A |
The heart of the PolF mechanism lies in its diiron centre's ability to activate molecular oxygen.
- O₂ Binding and μ-Peroxo Intermediate Formation: The diiron(II) centre in PolF activates O₂, forming a μ-peroxo-Fe(III)₂ intermediate [5]. This peroxo-bridged species is a potent oxidant.
- Unactivated C–H Bond Cleavage: Spectroscopic studies confirm that this μ-peroxo-Fe(III)â‚‚ intermediate is directly responsible for the cleavage of an unactivated C–H bond on the substrate [5]. This is a critical step, as the bond dissociation energy for such aliphatic C–H bonds can be as high as 101 kcal molâ»Â¹ [5]. This abstraction generates a substrate radical.
Radical-Mediated C–N Bond Formation and Cyclization
Following hydrogen abstraction, the reaction proceeds through a radical pathway to form the crucial C–N bond.
- Radical Rearrangement and Nitrogen Capture: The carbon-centred radical generated at C4 (from the numbering of the original amino acid) is poised for cyclization. The mechanism proposes that this radical attacks the lone pair on the nitrogen atom of the α-amino group [5]. This intramolecular radical addition results in the formation of a new C–N bond, creating a three-membered aziridine ring intermediate (e.g., 3-dimethylaziridine-2-carboxylic acid, Azi, was detected as an intermediate from L-Val) [5].
- Ring Expansion to Azetidine: The highly strained aziridine intermediate then undergoes a ring expansion. This step likely involves a second radical-based process, potentially another hydrogen abstraction or radical recombination, to form the more stable, though still strained, four-membered azetidine ring (e.g., MAA from L-Val) [5]. The crystal structure of PolF in complex with L-Ile provided critical insights into the spatial arrangement of the substrate that facilitates this radical cyclization [5] [11].
Table 3: Kinetic Profile of Observed Intermediates in PolF Single-Turnover Reaction with L-Val
| Observed Intermediate | Proposed Role in the Pathway | Relative Abundance / Kinetics |
|---|---|---|
| 3,4-dehydrovaline (3,4-dh-Val) | Initial desaturation product, direct precursor for cyclization [5]. | Major initial product (Formation rate: 0.23 minâ»Â¹) [5]. |
| 3-Dimethylaziridine-2-carboxylic acid (Azi) | Key cyclic intermediate, demonstrates C-N bond formation capability [5]. | Secondary product (Formation rate: 0.15 minâ»Â¹) [5]. |
| 4-Hydroxyvaline (4-OH-Val) | Off-pathway hydroxylation product [5]. | Secondary product (Formation rate: 0.14 minâ»Â¹) [5]. |
| 3-Hydroxyvaline (3-OH-Val) | Off-pathway hydroxylation product [5]. | Minor product (Formation rate: 0.036 minâ»Â¹) [5]. |
| 3-Methylene-azetidine-2-carboxylic acid (MAA) | Final azetidine product [5]. | Final product from 3,4-dh-Val conversion [5]. |
The elucidation of the PolE-PolF pathway provides a fundamental advance in our understanding of azetidine biosynthesis. It establishes a new paradigm for the construction of this strained ring system using non-haem iron enzymes, specifically through a mechanism involving substrate desaturation followed by a radical-mediated C–N bond formation catalyzed by an HDO superfamily enzyme [5]. This pathway is both energetically more efficient and biochemically distinct from previously known routes. For researchers and drug development professionals, these findings open up significant new avenues. The detailed mechanistic knowledge and the identification of the key intermediates enable the targeted engineering of these enzymes for biocatalytic applications. This could lead to more sustainable and efficient production of azetidine-based pharmaceuticals and fine chemicals, leveraging the power of enzymatic catalysis to overcome the traditional synthetic challenges associated with strained ring systems.
The biosynthesis of azetidine-containing amino acids represents a significant area of research in natural product chemistry and enzymology. The four-membered azetidine ring, characterized by substantial ring strain and unique reactivity, is a crucial structural motif in numerous bioactive compounds and pharmaceuticals [5] [7]. Within the broader context of biosynthesis research on azetidine amino acids by non-haem iron enzymes, understanding substrate specificity is fundamental for elucidating catalytic mechanisms and developing biocatalytic applications. This technical guide examines the substrate scope and transformation specificities of key non-haem iron-dependent enzymes, particularly PolF, which catalyzes the formation of azetidine rings from linear amino acid precursors.
The enzymatic machinery responsible for azetidine formation has remained enigmatic until recent discoveries identified specific non-haem iron enzymes capable of constructing these strained ring systems [5] [7]. Unlike traditional chemical synthesis that often requires harsh conditions and complex protection strategies, these enzymes achieve azetidine formation under mild physiological conditions with remarkable selectivity [7]. The focus on L-isoleucine and L-valine transformations stems from their role as natural substrates in the polyoxin antifungal pathway, where they are converted to azetidine-containing amino acids through novel oxidative mechanisms [5].
Enzymatic Transformations of Proteinogenic Amino Acids
PolF-Catalyzed Azetidine Ring Formation
PolF, identified as a member of the haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily, demonstrates remarkable capability in transforming aliphatic amino acids into azetidine derivatives [5]. This non-haem iron-dependent enzyme incorporates two iron atoms in its active site and utilizes molecular oxygen to initiate a series of oxidative transformations that ultimately lead to azetidine ring formation.
Genetic experiments involving gene knockout studies in Streptomyces cacaoi demonstrated that PolF is absolutely essential for polyoximic acid production, with polF mutants producing less than 1% of wild-type polyoxin A levels [5]. In vitro assays with purified PolF confirmed its catalytic function, showing direct conversion of L-isoleucine to polyoximic acid when supplemented with Fe(II) and O₂ [5]. This transformation proceeds via a 3,4-desaturated intermediate through a radical mechanism facilitated by a μ-peroxo-Fe(III)₂ intermediate that enables unactivated C–H bond cleavage [5].
Substrate Specificity Profile of PolF
The substrate scope of PolF has been systematically investigated using proteinogenic amino acids, revealing distinct preferences for specific aliphatic substrates. The enzyme demonstrates highest activity toward medium-size aliphatic amino acids with specific structural features that facilitate the cyclization process.
Table 1: Substrate Scope and Specificity of PolF
| Substrate | Product Formed | Type of Modification | Relative Conversion Efficiency | Key Structural Features |
|---|---|---|---|---|
| L-isoleucine | Polyoximic acid | Azetidine formation (-4 Da) | High | β-methyl group essential |
| L-valine | 3-methylene-azetidine-2-carboxylic acid (MAA) | Azetidine formation (-4 Da) | High | β-methyl group essential |
| L-leucine | Hydroxylation products (major), Minor desaturation | Hydroxylation (+16 Da) | Low | Lack of β-methyl group |
| L-methionine | Hydroxylation products (major), Minor desaturation | Hydroxylation (+16 Da) | Low | Sulfur-containing side chain |
| L-allo-isoleucine | Azetidine product | Azetidine formation (-4 Da) | Low | Stereochemistry at C2, C3 important |
| D-isoleucine | Azetidine product | Azetidine formation (-4 Da) | Low | Stereochemistry important |
| D-allo-isoleucine | Azetidine product | Azetidine formation (-4 Da) | Low | Stereochemistry important |
| Other proteogenic amino acids | No detectable products | - | None | - |
The data indicate that PolF exhibits strict requirement for β-methyl substitution, as evidenced by the efficient conversion of L-isoleucine and L-valine to azetidine products, while L-leucine and L-methionine primarily undergo hydroxylation with minimal azetidine formation [5]. Stereochemical preferences, though significant, are not absolute, as demonstrated by the low but detectable activity toward various isoleucine stereoisomers [5].
Mechanistic Insights into Azetidine Formation
Reaction Intermediates and Pathway
The mechanism of azetidine formation by PolF involves multiple oxidative steps with distinct intermediates. Single-turnover experiments with L-valine as substrate revealed several intermediates that provide crucial insight into the catalytic pathway:
Table 2: Reaction Intermediates in PolF-Catalyzed Azetidine Formation
| Intermediate | Structural Identification | Formation Rate (minâ»Â¹) | Role in Azetidine Pathway |
|---|---|---|---|
| 3,4-dehydrovaline (3,4-dh-Val) | Desaturated intermediate | 0.23 | Primary intermediate |
| 3-dimethylaziridine-2-carboxylic acid (Azi) | Aziridine ring | 0.15 | Potential branching intermediate |
| 4-hydroxyvaline (4-OH-Val) | Hydroxylated derivative | 0.14 | Competing side product |
| 3-hydroxyvaline (3-OH-Val) | Hydroxylated derivative | 0.036 | Competing side product |
| 3-methylene-azetidine-2-carboxylic acid (MAA) | Azetidine product | - | Final product |
Under multiple turnover conditions, 3,4-dehydrovaline is quantitatively converted to MAA, confirming its role as the key intermediate in azetidine formation [5]. The detection of an aziridine intermediate (Azi) suggests potential mechanistic parallels between three-membered and four-membered azacycle formation.
Catalytic Mechanism of C–N Bond Formation
PolF employs a unique radical mechanism for C–N bond formation that distinguishes it from other azetidine-forming enzymes. The catalytic cycle begins with the activation of molecular oxygen by the diiron center, generating a μ-peroxo-Fe(III)₂ intermediate that abstracts hydrogen from the substrate [5]. This initial H-abstraction is followed by a series of radical reactions that ultimately lead to C–N bond formation and azetidine ring closure.
The enzyme's ability to catalyze three distinct reactions—desaturation, hydroxylation, and C–N bond formation—on a single substrate highlights its remarkable catalytic versatility [5]. The preference for azetidine formation with specific substrates suggests that the enzyme active site precisely orients the radical intermediate to favor intramolecular C–N bond formation over other possible outcomes.
Figure 1: Proposed Mechanism for PolF-Catalyzed Azetidine Formation from L-Isoleucine
Experimental Protocols for Studying Substrate Specificity
Enzyme Purification and Reconstitution
PolF Expression and Purification:
- Express PolF in E. coli host system [5]
- Purify using affinity chromatography under anaerobic conditions [5]
- Isolate initially in apo form (largely iron-free) [5]
Iron Reconstitution:
- Incubate apo-PolF with excess Fe(II) (3 equivalents) under anaerobic conditions [5]
- Remove unbound Fe(II) using desalting column [5]
- Typical iron content after reconstitution: ~1.5 equivalents per enzyme [5]
- Note: Less than 2 equivalents Fe bound is consistent with weak Fe affinity in HDO enzymes without substrate [5]
Enzyme Activity Assays
Standard Reaction Conditions:
- Prepare enzyme and substrate mixture anaerobically [5]
- Initiate reaction by adding Oâ‚‚-saturated buffer [5]
- Essential components: Reconstituted PolF, amino acid substrate, Fe(II), Oâ‚‚ [5]
- Include external reductant (ascorbate or dithiothreitol) for multiple turnovers [5]
Product Analysis:
- Derivatize reaction products with dansyl chloride (DnsCl) [5]
- Analyze by liquid chromatography-mass spectrometry (LC-MS) [5]
- Identify products by molecular weight changes (-4 Da for azetidine products) [5]
- Confirm structures by comparison with authentic standards and NMR characterization [5]
Single Turnover Conditions:
- Use 2 equivalents Fe(II) with no external reductant [5]
- Allows detection of reaction intermediates [5]
- Measure formation rates of different products [5]
Substrate Specificity Screening
Comprehensive Screening Approach:
- Test all 20 proteogenic amino acids individually [5]
- Include stereoisomers (L-allo-Ile, D-Ile, D-allo-Ile) [5]
- Analyze products for modifications: -4 Da (azetidine), -2 Da (desaturation), +16 Da (hydroxylation) [5]
- Use commercial standards when available for product identification [5]
Auxiliary Enzymes and Pathway Optimization
Role of PolE in Enhancing Biosynthetic Flux
While PolF alone is sufficient for azetidine formation, the polyoxin biosynthetic pathway includes PolE, a member of the DUF6421 family, which enhances the efficiency of the process [5]. PolE functions as an iron and pterin-dependent oxidase that specifically catalyzes the desaturation of L-isoleucine, creating the 3,4-desaturated intermediate that PolF utilizes more efficiently than the saturated amino acid [5].
Genetic evidence supporting this auxiliary role comes from gene knockout experiments, where polE disruption reduced polyoxin A production to approximately 10% of wild-type levels, compared to complete abolition with polF disruption [5]. This indicates that while PolE is not absolutely essential for azetidine formation, it significantly increases biosynthetic flux by providing a more suitable substrate for PolF.
Figure 2: Biosynthetic Pathway for Polyoximic Acid Formation Showing PolE and PolF Roles
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Studying Azetidine Amino Acid Biosynthesis
| Reagent/Chemical | Function/Application | Specific Usage Notes |
|---|---|---|
| Purified PolF enzyme | Catalytic core component | Express in E. coli; purify under anaerobic conditions |
| Fe(II) salts (e.g., FeSOâ‚„) | Cofactor for PolF | Use 3 eq. for reconstitution; anaerobic handling critical |
| Oâ‚‚-saturated buffer | Oxidant for reaction | Saturate buffer with Oâ‚‚ prior to reaction initiation |
| Ascorbate/DTT | External reductant | Essential for multiple turnover conditions |
| L-isoleucine | Natural substrate | Primary substrate for polyoximic acid production |
| L-valine | Alternative substrate | Forms 3-methylene-azetidine-2-carboxylic acid |
| Dansyl chloride (DnsCl) | Derivatization agent | Enables LC-MS detection and analysis of products |
| Polyoxin A standard | Authentic standard | Source of polyoximic acid for comparison |
| 3,4-dehydrovaline standard | Intermediate standard | For identification of reaction intermediate |
| (S)-(+)-N-3-Benzylnirvanol | (S)-(+)-N-3-Benzylnirvanol, CAS:790676-40-3, MF:C18H18N2O2, MW:294.3 g/mol | Chemical Reagent |
| 2-Ethyl-2-phenylmalonamide-d5 | 2-Ethyl-2-phenylmalonamide-d5, MF:C11H14N2O2, MW:211.27 g/mol | Chemical Reagent |
Implications for Pharmaceutical Development
The elucidation of substrate scope and specificity for azetidine-forming enzymes has significant implications for drug discovery and development. Azetidine rings are prized in medicinal chemistry for their ability to modulate molecular rigidity, improve metabolic stability, and enhance target binding affinity [7]. Understanding the natural biosynthetic pathways for these strained rings provides opportunities for biocatalytic production of azetidine-containing pharmaceutical intermediates.
The substrate flexibility observed with PolF, particularly its ability to process both L-isoleucine and L-valine into azetidine products, suggests potential for engineering modified enzymes with altered specificity profiles. Such engineered enzymes could enable biosynthesis of novel azetidine compounds not found in nature, expanding the chemical space available for drug discovery programs [5] [7].
Furthermore, the mechanistic insights gained from studying PolF's radical-based C–N bond formation may inspire development of biomimetic synthetic methodologies for azetidine ring construction, potentially overcoming current limitations in chemical synthesis of these strained heterocycles [5].
Azetidines, four-membered nitrogen-containing saturated heterocycles, represent a crucial structural motif in numerous bioactive compounds and pharmaceutical agents due to their unique three-dimensional shape and significant ring strain (approximately 25.4 kcal molâ»Â¹) [5]. This inherent strain contributes to their exceptional reactivity and biological activity, making them valuable building blocks in drug development [7]. Despite their pharmaceutical importance, the synthesis of azetidine-containing compounds has presented formidable challenges for synthetic chemists, traditionally requiring harsh solvents, toxic chemicals, and complex multi-step processes that generate substantial waste [23]. The inherent difficulty stems from the need to bend chemical bonds at acute angles to form the strained four-membered ring system, a process that typically demands significant energy input and specialized conditions [28].
Within this challenging landscape, biocatalytic strategies have emerged as transformative alternatives, offering sustainable pathways for azetidine synthesis under mild, environmentally friendly conditions [7]. Nature has evolved sophisticated enzymatic machinery capable of constructing these strained ring systems with remarkable precision and efficiency. Recent groundbreaking research has illuminated previously enigmatic biosynthetic pathways, particularly those mediated by non-haem iron-dependent enzymes, which catalyze the formation of azetidine rings from simple amino acid precursors [5] [6]. These biological routes operate in aqueous environments at ambient temperatures and pressures, bypassing the need for expensive protecting groups and hazardous reagents typically employed in traditional synthetic approaches [23].
The discovery and characterization of specialized enzymes such as PolF and PolE from the polyoxin biosynthetic pathway represent significant advances in the field of sustainable synthesis [5]. These non-haem iron-dependent enzymes demonstrate nature's ability to perform chemically challenging transformations, including unactivated C–H bond cleavage and subsequent C–N bond formation, through radical mechanisms that avoid the environmental burdens associated with conventional synthetic methods [5] [7]. By harnessing and engineering these biological catalysts, researchers can now access azetidine scaffolds with greater efficiency and selectivity while significantly reducing the ecological footprint of synthetic processes.
Enzymatic Machinery for Azetidine Formation
Key Enzymes and Mechanisms
The biosynthesis of azetidine amino acids involves specialized enzymatic machinery that catalyzes the formation of strained four-membered rings through unique mechanisms. Research has revealed two distinct enzymatic strategies for azetidine ring formation in nature, each employing different cofactors and catalytic approaches.
Table 1: Key Enzymes in Azetidine Biosynthesis
| Enzyme | Class | Cofactor | Reaction Catalyzed | Natural Product Association |
|---|---|---|---|---|
| PolF | HDO superfamily | Non-haem diiron | Azetidine formation from L-Ile/L-Val via desaturation | Polyoximic acid in polyoxin antifungal compounds |
| PolE | DUF6421 family | Iron and pterin | Desaturation of L-Ile | Polyoximic acid in polyoxin |
| AzeJ/VioH | MT1 family | None (SAM-dependent) | SAM cyclization to AZE | Azetidomonamides and vioprolides |
The PolF enzyme, a member of the haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily, represents a particularly remarkable biocatalyst [5]. This non-haem iron-dependent enzyme alone is sufficient to transform proteinogenic amino acids L-isoleucine (L-Ile) and L-valine (L-Val) into their corresponding azetidine derivatives via a 3,4-desaturated intermediate [5] [6]. Genetic knockout experiments in Streptomyces cacaoi demonstrated that PolF is absolutely essential for polyoximic acid biosynthesis, with the polF mutant producing no measurable polyoxin A (<1% of wild-type) [5]. Mechanistic studies indicate that PolF employs a μ-peroxo-Fe(III)â‚‚ intermediate to directly cleave unactivated C–H bonds (with bond strengths up to 101 kcal molâ»Â¹), with subsequent C–N bond formation proceeding through radical mechanisms [5].
Simultaneously, a distinct biosynthetic pathway employs SAM-dependent AZE synthases such as AzeJ and VioH, which catalyze the intramolecular 4-exo-tet cyclization of S-adenosylmethionine (SAM) to yield azetidine-2-carboxylic acid (AZE) [29]. Structural analyses of these enzymes reveal that cyclization is facilitated by an exceptional substrate conformation supported by desolvation effects and cation-Ï€ interactions [29]. These enzymes belong to the class I methyltransferase (MT1) family but have evolved to promote cyclization rather than methylation, highlighting nature's ability to repurpose protein folds for divergent functions [29].
The following diagram illustrates the primary enzymatic pathways for azetidine biosynthesis:
Diagram 1: Enzymatic Pathways for Azetidine Biosynthesis. This diagram illustrates the two primary biological routes to azetidine scaffolds: the PolE/PolF pathway from proteinogenic amino acids and the SAM-dependent cyclization pathway.
Structural Features and Catalytic Innovations
The structural biology of azetidine-forming enzymes reveals remarkable adaptations for catalyzing challenging ring-forming reactions. PolF incorporates a diiron center within a protein scaffold that facilitates unique reactivity profiles specifically tailored for ring formation [7]. Structural analysis using X-ray crystallography shows that the enzyme's active site positions substrates precisely to enable the geometrically constrained transition state required for four-membered ring formation [5]. The iron(IV)-oxo intermediate generated during catalysis abstracts hydrogen atoms and facilitates intramolecular C–N bond formation through radical mechanisms that would be difficult to control in conventional synthetic chemistry [7].
For the SAM-dependent AZE synthases, structural studies reveal that substrate conformation is critical for cyclization [29]. The homocysteine moiety of SAM adopts a kinked conformation that directs the nucleophilic nitrogen toward the Cγ-carbon (2.9 Å distance), enabling the 4-exo-tet cyclization that would be disfavored in solution [29]. This kinked conformation is stabilized by specific protein interactions, including hydrogen bonding between the HCY-COO⻠and Tyr176, Asn139, and Ser203, as well as a π-interaction between Phe134 and HCY-NH₂ that increases nitrogen nucleophilicity [29]. These structural features work in concert to overcome the inherent geometric constraints of forming small rings, showcasing nature's sophisticated solutions to challenging chemical transformations.
Experimental Protocols for Enzyme Characterization
Enzyme Production and Purification
The functional characterization of azetidine biosynthetic enzymes begins with recombinant production and purification. For PolF and PolE studies, researchers typically express these enzymes heterologously in E. coli systems [5]. The protocol involves transforming E. coli BL21(DE3) cells with plasmid vectors containing the genes of interest (e.g., polF or polE), followed by growth in LB medium supplemented with appropriate antibiotics. Protein expression is induced by adding isopropyl β-D-1-thiogalactopyranoside (IPTG) when cultures reach an optical density (OD₆₀₀) of approximately 0.6-0.8, followed by continued incubation at 16-18°C for 16-20 hours to promote proper folding [5].
Cell harvesting via centrifugation is followed by lysis using sonication or French press in a suitable buffer (typically 50 mM HEPES, pH 7.5, containing 300 mM NaCl). The crude lysate is clarified by centrifugation, and the supernatant is subjected to immobilized metal affinity chromatography (IMAC) using nickel-nitrilotriacetic acid (Ni-NTA) resin, exploiting engineered hexahistidine tags on the recombinant proteins [5]. After washing with buffer containing 20-50 mM imidazole, the target proteins are eluted with buffer containing 250-300 mM imidazole. Further purification is achieved through size-exclusion chromatography (SEC) using columns such as Superdex 200, equilibrated with storage buffer (e.g., 25 mM HEPES, pH 7.5, 150 mM NaCl) [5]. Protein purity is assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and concentrations are determined by Bradford assay or ultraviolet (UV) absorbance at 280 nm.
Enzyme Activity Assays
Activity assays for azetidine-forming enzymes require careful handling due to the oxygen-sensitive nature of their metallocenters. For PolF, researchers first generate apo-enzyme by chelating bound metals, then reconstitute active enzyme by incubating apo-PolF with excess Fe(II) (typically 3 equivalents) under anaerobic conditions in an inert atmosphere glovebox [5]. Excess metal is removed using desalting columns, with metal incorporation quantified by atomic absorption spectroscopy or colorimetric assays [5].
Standard reaction mixtures contain reconstituted enzyme (50-100 μM), substrate (L-Ile or L-Val, 1-5 mM), and ascorbate or dithiothreitol (1-5 mM) as an external reductant in anaerobic buffer [5]. Reactions are initiated by adding O₂-saturated buffer and proceed at 25-30°C with shaking. For time-course experiments, aliquots are removed at intervals and quenched with acid or organic solvent. Reaction products are typically derivatized with dansyl chloride (DnsCl) for detection and analyzed by liquid chromatography-mass spectrometry (LC-MS) [5]. For the SAM-dependent AZE synthases, assays monitor the conversion of SAM to AZE and methylthioadenosine (MTA) using high-performance liquid chromatography (HPLC) or LC-MS, with kinetic parameters determined by varying SAM concentration [29].
Table 2: Key Reagents for Azetidine Biosynthesis Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Enzyme Expression | E. coli BL21(DE3), pET vectors, IPTG | Heterologous protein production |
| Purification | Ni-NTA resin, Imidazole, Size-exclusion columns | Protein isolation and purification |
| Activity Assays | L-Ile, L-Val, SAM, Sodium ascorbate, DTT | Enzyme substrates and redox cofactors |
| Analytical Methods | DnsCl, LC-MS, HPLC, Mössbauer spectroscopy | Product derivatization, detection, and characterization |
| Specialized Equipment | Anaerobic chamber, Stopped-flow spectrometer | Handling oxygen-sensitive enzymes and intermediates |
Spectroscopic Characterization
Advanced spectroscopic techniques provide crucial insights into the metallocenters and reactive intermediates of azetidine biosynthetic enzymes. For PolF, stopped-flow spectroscopy combined with Mössbauer spectroscopy reveals the formation of a μ-peroxo-Fe(III)₂ intermediate responsible for the challenging C–H bond cleavage [5] [23]. These experiments require specialized equipment for rapid mixing and freezing of samples at specific time points (e.g., 50-500 milliseconds after reaction initiation) [5]. Mössbauer spectra are typically recorded at 4.2 K in a magnetic field, with data analysis providing information on iron oxidation states and coordination environments [5].
X-ray crystallography remains an essential tool for determining enzyme structures. Crystals of AzeJ and VioH have been obtained by the sitting-drop vapor diffusion method at 20°C, mixing protein solution (10-15 mg/mL in 20 mM HEPES, pH 7.5, 150 mM NaCl) with reservoir solution containing 20-25% PEG 3350 and 0.2 M ammonium acetate [29]. For ligand-bound structures, enzymes are co-crystallized with substrates (SAM) or products (SAH, AZE). Diffraction data collected at synchrotron sources (e.g., 1.95 Å resolution for AzeJ-SAM complex) enables visualization of active site architecture and substrate positioning [29].
Biocatalytic Applications and Advantages
Sustainable Synthesis of Pharmaceutical Intermediates
The implementation of enzymatic routes for azetidine synthesis offers substantial advantages over conventional chemical methods in terms of sustainability, efficiency, and selectivity. Traditional chemical synthesis of azetidine scaffolds often requires multi-step sequences, expensive catalysts, harsh reagents, and protected precursors, resulting in significant waste generation and environmental impact [23]. In contrast, biocatalytic approaches employing enzymes like PolF and AzeJ accomplish these challenging transformations in single steps under mild aqueous conditions, with molecular oxygen as the primary oxidant [5] [7].
The economic benefits are particularly noteworthy. Previously reported enzymatic processes for producing azetidine required expensive precursor compounds, with one supplier charging approximately $1,390 per gram [23]. The newly discovered enzymes utilize inexpensive precursor compounds that cost roughly 1,000-fold less, dramatically reducing production costs for these valuable pharmaceutical intermediates [23]. Furthermore, the operational simplicity of biocatalytic processes—conducted at ambient temperature and pressure in water—translates to reduced energy consumption and lower capital investment in specialized equipment capable of withstanding extreme conditions [23] [7].
The following diagram illustrates a generalized workflow for biocatalytic azetidine production:
Diagram 2: Workflow for Biocatalytic Azetidine Production. This diagram outlines the general process for enzymatic synthesis of azetidines, highlighting the use of inexpensive precursors and mild aqueous conditions.
Substrate Scope and Enzyme Versatility
Biocatalytic routes to azetidines demonstrate remarkable versatility in substrate acceptance. PolF exhibits selective activity toward medium-size aliphatic amino acids, efficiently converting L-Ile and L-Val to their corresponding azetidine derivatives while showing minimal activity toward other proteinogenic amino acids [5]. Interestingly, PolF also accepts stereoisomers of isoleucine (L-allo-Ile, D-Ile, and D-allo-Ile), producing azetidine products in detectable amounts, albeit with reduced efficiency compared to the natural L-Ile substrate [5]. This relaxed stereospecificity expands the structural diversity of accessible azetidine compounds.
Remarkably, PolF demonstrates catalytic promiscuity beyond azetidine formation, capable of producing an even smaller compound, aziridine, which features a nitrogen-containing three-membered ring [23]. This ability to form both four-membered and three-membered nitrogen heterocycles is unique and unprecedented in enzymatic catalysis, highlighting the potential for discovering multifunctional biocatalysts from natural biosynthetic pathways [23]. Meanwhile, SAM-dependent AZE synthases like AzeJ exhibit strict substrate specificity for SAM but can be engineered through protein engineering approaches to accept analogs with modified amino acid side chains, further expanding the range of accessible azetidine derivatives [29].
Future Directions and Implementation Strategies
Enzyme Engineering and Optimization
The future development of azetidine biocatalysis will heavily rely on advanced enzyme engineering strategies to enhance catalytic efficiency, substrate scope, and operational stability. Structure-guided mutagenesis based on high-resolution crystal structures of AzeJ (1.95 Ã… resolution) and PolF enables targeted modifications of active site residues to improve substrate binding or alter reaction selectivity [29]. For instance, residues involved in coordinating the homocysteine moiety of SAM in AzeJ (Tyr175, Tyr176, Asn139, Ser203) represent potential targets for engineering altered substrate specificity [29].
Directed evolution approaches employing iterative rounds of random mutagenesis and screening offer powerful complementary strategies for improving biocatalyst performance without requiring detailed structural information [7]. The combination of these methods with machine learning algorithms that predict function-enhancing mutations based on sequence-activity relationships will accelerate the development of optimized azetidine-forming enzymes [7]. Furthermore, fusion protein strategies that covalently link collaborating enzymes like PolE and PolF could enhance metabolic channeling and overall pathway efficiency by increasing the local concentration of intermediates [5].
Industrial Implementation and Scale-Up
The transition from laboratory-scale biocatalysis to industrial implementation requires addressing several practical considerations, including enzyme immobilization for reusability, cofactor regeneration systems, and integration with chemical synthesis steps. Robust immobilization supports such as epoxy-functionalized resins or magnetic nanoparticles can significantly enhance operational stability and enable repeated batch reactions or continuous flow processes [30]. The development of efficient cofactor regeneration systems is particularly important for NADPH-dependent enzymes like PolE, which may be addressed through substrate-coupled approaches or enzymatic regeneration systems [5].
For large-scale production, continuous flow bioreactors offer advantages over traditional batch processes, including improved mass transfer, better temperature control, and easier product separation [30]. Recent advances have demonstrated the feasibility of continuous flow hydrogenation of 2-azetines using environmentally responsible solvents like ethyl acetate and cyclopentyl methyl ether (CPME), suggesting potential for integrating biocatalytic and chemical steps in telescoped multistep processes [30]. Such integrated approaches leverage the strengths of both biological and chemical catalysis while minimizing environmental impact and operational costs.
As biocatalytic methods for azetidine synthesis continue to mature, their implementation in pharmaceutical manufacturing will expand, driven by increasing regulatory pressure for sustainable processes and the growing demand for complex chiral intermediates. With ongoing advances in enzyme discovery, engineering, and process optimization, biocatalytic routes are poised to become the preferred methods for synthesizing these valuable strained heterocycles, ultimately contributing to greener and more efficient pharmaceutical production.
Overcoming Challenges in Enzymatic Azetidine Formation
The biosynthesis of azetidine amino acids represents a significant area of research in natural product chemistry and enzymology, particularly given the pharmaceutical relevance of these strained four-membered heterocycles. The recent discovery of a novel biosynthetic pathway in the polyoxin antifungal system, mediated by non-haem iron-dependent enzymes, provides a groundbreaking framework for understanding how nature constructs these challenging ring systems [5] [7]. This technical guide examines the comprehensive strategies employed for trapping and analyzing the key intermediates in the azetidine amino acid biosynthesis pathway, with particular focus on the enzymatic mechanisms of PolE and PolF enzymes. The methodologies outlined herein offer researchers a robust toolkit for investigating complex biosynthetic pathways involving high-energy intermediates and radical mechanisms.
Experimental Approaches for Intermediate Identification
Genetic and In Vivo Validation Strategies
Initial evidence for the involvement of specific enzymes in azetidine biosynthesis came from systematic genetic manipulation of the producing organism. In the polyoxin system, researchers performed in-frame deletion of polE and polF genes in Streptomyces cacaoi to establish their functional roles [5] [10]. The resulting mutants were cultured under polyoxin-producing conditions, and the fermentation broth was analyzed using liquid chromatography-mass spectrometry (LC-MS). This approach revealed that the polF mutant produced virtually no polyoxin A (<1% of wild-type), establishing PolF as essential for azetidine formation [5] [10]. The polE mutant showed reduced but detectable polyoxin A production (~10% of wild-type), suggesting a supplementary role in enhancing biosynthetic flux [5] [10]. This genetic validation provides a critical first step in establishing which enzymes warrant detailed mechanistic investigation for intermediate trapping studies.
In Vitro Enzymatic Assays and Intermediate Trapping
Protein Expression and Purification: PolF was expressed in E. coli and purified, initially obtaining the enzyme in predominantly apo form [5] [10]. Since haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) family members require two equivalents of Fe(II), the apo-PolF was reconstituted by incubation with excess Fe(II) (3 equivalents) under anaerobic conditions [5] [10]. After removing unbound Fe(II) using a desalting column, the resulting PolF contained approximately 1.5 equivalents of Fe(II), consistent with the weak iron affinity reported for HDO enzymes in the absence of substrate [5] [10].
Activity Assay Conditions: Standard assays contained PolF reconstituted with Fe(II), substrate (L-isoleucine or L-valine), and were prepared anaerobically [5] [10]. Reactions were initiated by adding Oâ‚‚-saturated buffer. For multiple turnover conditions, external reductants such as ascorbate or dithiothreitol were included to maintain the diiron center in its reduced state during catalytic cycling [5] [10]. Single-turnover conditions utilized only 2 equivalents of Fe(II) with no external reductant, allowing observation of initial reaction products without further turnover [5].
Intermediate Analysis: Reaction products were derivatized with dansyl chloride (DnsCl) to enhance detection sensitivity and analyzed by LC-MS [5] [10]. This approach enabled identification of products with characteristic mass changes, including -4 Da (azetidine products), -2 Da (desaturation products), and +16 Da (hydroxylation products) relative to the substrate [5].
Strategic Use of Alternative Substrates
The investigation of PolF's substrate specificity provided critical insights into the biosynthetic mechanism and enabled the trapping of intermediates that were less abundant with the native substrate. When researchers tested L-valine as an alternative substrate, they observed the formation of multiple intermediates that were characterized as 3-hydroxyvaline (3-OH-Val), 4-hydroxyvaline (4-OH-Val), 3,4-dehydrovaline (3,4-dh-Val), and 3-dimethylaziridine-2-carboxylic acid (Azi) [5]. The comparative abundance of these intermediates with L-valine, as opposed to L-isoleucine, facilitated mechanistic proposals regarding the azetidine formation pathway.
Table 1: Key Intermediates Identified in PolF-Catalyzed Azetidine Formation
| Intermediate | Mass Change | Identification Method | Proposed Role in Pathway |
|---|---|---|---|
| 3,4-dehydrovaline (3,4-dh-Val) | -2 Da | Comparison to authentic standard | Central desaturated intermediate |
| 3-hydroxyvaline (3-OH-Val) | +16 Da | Comparison to authentic standard | Hydroxylation side product |
| 4-hydroxyvaline (4-OH-Val) | +16 Da | Comparison to authentic standard | Hydroxylation side product |
| 3-dimethylaziridine-2-carboxylic acid (Azi) | -4 Da | NMR characterization | Aziridine intermediate |
| 3-methylene-azetidine-2-carboxylic acid (MAA) | -4 Da | NMR characterization | Final azetidine product from L-valine |
Structural Biology Approaches
X-ray crystallography provided critical structural information for understanding the mechanism of azetidine formation. Researchers determined the crystal structure of PolF in complex with L-Ile, revealing the substrate binding mode and active site architecture [5] [11]. This structural information enabled precise proposals regarding how the enzyme positions the substrate for C-H activation and subsequent C-N bond formation, and informed understanding of the radical rearrangement mechanism [5] [11].
Biosynthetic Pathway and Intermediate Trapping Workflow
The following diagram illustrates the integrated experimental workflow for identifying and analyzing key intermediates in the azetidine biosynthetic pathway:
Key Intermediates and Mechanistic Insights
The Central Desaturated Intermediate
A critical breakthrough in understanding the azetidine biosynthesis pathway came from the identification of 3,4-dehydro-isoleucine as a key intermediate [5]. This desaturated species is generated through the action of PolE, an Fe- and pterin-dependent oxidase that enhances the flux through the azetidine formation pathway by providing the desaturated substrate to PolF [5] [10]. Under single-turnover conditions, 3,4-dehydrovaline emerged as the major initial product when L-valine was used as substrate, appearing with a formation rate of 0.23 minâ»Â¹ [5]. This intermediate was quantitatively converted to the azetidine product when provided to PolF, establishing its role as a direct precursor in the pathway [5].
Unexpected Aziridine Intermediate
A significant finding from the intermediate trapping studies was the identification of an aziridine-containing compound, 3-dimethylaziridine-2-carboxylic acid (Azi), during PolF catalysis with L-valine [5]. This three-membered nitrogen heterocycle represented the first enzymatic example of C-N bond formation in the pathway and appeared with a formation rate of 0.15 minâ»Â¹ under single-turnover conditions [5]. The discovery of this intermediate provided strong evidence for a stepwise mechanism in which C-N bond formation precedes ring expansion to the azetidine, contrary to potential alternative mechanisms involving direct cyclization.
Hydroxylated Byproducts as Mechanistic Probes
The trapping of hydroxylated products (3-OH-Val and 4-OH-Val) provided additional evidence for the radical nature of the mechanism [5]. These products likely result from radical quenching before C-N bond formation, with formation rates of 0.036 minâ»Â¹ and 0.14 minâ»Â¹, respectively [5]. The relative abundance of 4-OH-Val compared to 3-OH-Val suggests that the C4 position may be the initial site of hydrogen atom abstraction, consistent with the predicted bond dissociation energies and the stability of the resulting radical intermediate.
The following diagram illustrates the mechanistic pathway and key intermediates in the PolF-catalyzed azetidine formation:
Research Reagent Solutions for Intermediate Trapping
Table 2: Essential Research Reagents for Azetidine Biosynthesis Studies
| Reagent | Function/Application | Experimental Considerations |
|---|---|---|
| PolF Enzyme (HDO family) | Catalyzes azetidine formation from L-Ile/L-Val | Requires reconstitution with Fe(II); exhibits weak iron affinity without substrate [5] [10] |
| PolE Enzyme (DUF6421 family) | Fe- and pterin-dependent oxidase that desaturates L-Ile | Enhances flux through pathway by providing desaturated intermediate [5] [10] |
| Fe(II) (e.g., FeSOâ‚„) | Cofactor for PolF and PolE | Must be added anaerobically; 3 equivalents used for reconstitution [5] [10] |
| Ascorbate/DTT | External reductant for multiple turnover conditions | Maintains diiron center in reduced state during catalysis [5] |
| Dansyl Chloride (DnsCl) | Derivatizing agent for LC-MS analysis | Enhances detection sensitivity of amino acid products [5] [10] |
| Oâ‚‚-saturated buffer | Oxidant for initiating reactions | Added to initiate catalysis after anaerobic enzyme-substrate mixing [5] [10] |
| 3,4-dehydrovaline standard | Authentic standard for intermediate identification | Enables confirmation of desaturated intermediate identity [5] |
Spectroscopic Characterization of Reactive Species
Mechanistic studies of PolF provided evidence for a μ-peroxo-Fe(III)₂ intermediate that is directly responsible for the unactivated C-H bond cleavage [5] [10]. This high-valent diiron species represents the key oxidizing agent in the reaction, capable of abstracting hydrogen atoms from strong C-H bonds with dissociation energies up to 101 kcal/mol [10]. Spectroscopic approaches, including analysis of the Fe₂ cluster, supported the formation of this intermediate and its role in the radical mechanism [5] [11]. The post-hydrogen-abstraction reactions, including the crucial C-N bond formation step, were shown to proceed through radical mechanisms based on the trapping of radical rearrangement products and characterization of the aziridine intermediate [5].
The strategies for trapping and analyzing key intermediates in azetidine amino acid biosynthesis have revealed unprecedented enzymatic mechanisms for constructing strained heterocyclic systems. The combination of genetic approaches, in vitro enzymology with carefully controlled turnover conditions, strategic use of alternative substrates, and structural biology has provided a comprehensive understanding of how non-haem iron enzymes work in concert to build the azetidine ring through a radical-based mechanism. These methodologies offer a template for investigating other enigmatic biosynthetic pathways and highlight the sophisticated approaches required to characterize transient intermediates in complex enzymatic transformations. The continued application and refinement of these strategies will undoubtedly lead to further discoveries in natural product biosynthesis and enable bioinspired approaches to synthetically challenging molecular architectures.
The biosynthesis of complex natural products often involves enzymatic steps that transform canonical amino acids into specialized structures with potent bioactivity. A compelling example is the formation of the azetidine ring—a strained, four-membered nitrogen-containing heterocycle found in the antifungal agent polyoxin A. This structure, known as polyoximic acid (PA), is derived from L-isoleucine (L-Ile) via a pathway that was, until recently, poorly understood [5] [10]. Within this pathway, the enzyme PolE plays a critical role by performing an initial key activation, thereby enhancing the overall flux through the biosynthetic route. This whitepaper details the function of PolE, its mechanistic basis, and its synergistic relationship with the cyclizing enzyme PolF, providing a comprehensive technical guide for researchers in enzymology and natural product biosynthesis.
PolE's Functional Role in the Azetidine Biosynthetic Pathway
Genetic and Functional Context within the Polyoxin Cluster
The polyoxin biosynthetic gene cluster in Streptomyces cacaoi contains several genes implicated in the formation of the azetidine amino acid. Early genetic knockout experiments established the essentiality of two genes, polE and polF, for efficient polyoxin production [5] [10]. While the inactivation of polF completely abolished polyoxin A production (yielding <1% of wild-type levels), the disruption of polE led to a significant but less severe reduction, with the mutant still producing approximately 10% of the wild-type titre [5]. This genetic evidence suggested that while PolF is absolutely indispensable for azetidine formation, PolE serves a supplementary function that substantially boosts the efficiency of the pathway.
Biochemical Function: Targeted Desaturation of L-Isoleucine
PolE is a member of the DUF6421 protein family and functions as a novel Fe and pterin-dependent oxidase [5] [10]. Its primary biochemical role is to catalyze the desaturation of L-Ile, specifically introducing a double bond between the C3 and C4 carbon atoms of its aliphatic side chain to form a 3,4-dehydroisoleucine intermediate (a -2 Da mass change) [5] [11]. This 3,4-desaturation is a crucial activation step that prepares the substrate for the subsequent cyclization reaction catalyzed by PolF.
Diagram 1: Enzymatic cascade for azetidine biosynthesis showing PolE's role in initial desaturation.
Mechanistic Basis for Flux Enhancement
Kinetic Facilitation of the Rate-Limiting Step
The transformation of L-Ile into the strained azetidine ring is chemically challenging. PolF alone can catalyze the complete multi-step transformation from L-Ile to PA, but its catalytic efficiency is significantly lower without the preparatory action of PolE [5] [10]. Under single-turnover conditions, the desaturation of L-Ile by PolF is a relatively slow step. By pre-forming the 3,4-dehydroisoleucine intermediate, PolE effectively bypasses this kinetic bottleneck, increasing the local concentration of this key intermediate and allowing PolF to operate more efficiently on a substrate primed for cyclization [5]. This division of labor enhances the overall flux through the pathway, ensuring robust production of the azetidine pharmacophore.
Substrate Specificity and Pathway Fidelity
PolE exhibits a defined substrate specificity that directs metabolic flux toward the correct biosynthetic product. The enzyme is specific for L-Ile, the physiological precursor for polyoximic acid [5]. This specificity ensures that the desaturation activity is channeled into the polyoxin pathway, maintaining the fidelity of the biosynthetic process and avoiding potential off-target reactions with other cellular amino acids.
Table 1: Comparative Substrate Specificity of PolE and PolF
| Enzyme | Primary Substrate(s) | Reaction Catalyzed | Key Cofactors | Product(s) |
|---|---|---|---|---|
| PolE | L-Isoleucine | 3,4-Desaturation | Fe²âº, Pterin | 3,4-Dehydroisoleucine |
| PolF | L-Ile, L-Val, 3,4-Dehydroisoleucine | Desaturation, Hydroxylation, C-N Cyclization | Diiron Center (μ-peroxo-Fe(III)₂) | Polyoximic Acid, MAA, Hydroxylated byproducts |
Experimental Characterization of PolE
In Vitro Enzyme Assay Protocol
The functional characterization of PolE requires a reconstituted enzyme system under controlled conditions.
- Enzyme Preparation: Heterologously express and purify PolE from E. coli. The enzyme is typically isolated in an apo form and requires reconstitution with Fe²⺠[5] [10].
- Reconstitution: Incubate apo-PolE with an excess of Fe(II) (e.g., 3 equivalents) under anaerobic conditions in an inert atmosphere glovebox to prevent premature oxidation. Remove unbound metal ions using a desalting column [5].
- Reaction Setup: Prepare the reaction mixture anaerobically by combining reconstituted PolE (e.g., 5-10 µM) with substrate L-Ile (e.g., 100-500 µM) in a suitable buffer.
- Reaction Initiation & Quenching: Initiate the reaction by introducing an Oâ‚‚-saturated buffer to start the oxidative desaturation. Allow the reaction to proceed for a set time (e.g., 30-60 minutes) before quenching, typically by acidification or rapid freezing [5].
- Product Analysis: Derivatize the reaction products with dansyl chloride (DnsCl) for analysis. Detect and quantify the 3,4-dehydroisoleucine product (-2 Da from L-Ile) using Liquid Chromatography-Mass Spectrometry (LC-MS). The identity of the product can be confirmed by comparison with authentic standards when available [5].
Key Analytical Findings and Data Interpretation
Application of the above protocol confirmed PolE's catalytic activity. LC-MS analysis revealed a product with a mass shift of -2 Da relative to L-Ile, consistent with a desaturation reaction [5]. Control experiments omitting Fe(II), Oâ‚‚, or using heat-inactivated enzyme resulted in no product formation, confirming the enzyme's dependence on both the metal cofactor and oxygen [5]. The activity of PolE was also found to be dependent on the presence of an external reductant, such as ascorbate or dithiothreitol, which is consistent with the need to maintain the iron cofactor in its reduced state for multiple catalytic turnovers [5].
Diagram 2: Experimental workflow for in vitro characterization of PolE activity.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Studying PolE and Azetidine Biosynthesis
| Reagent / Material | Function in Research | Specific Application Example |
|---|---|---|
| Apo-PolE Enzyme | Catalytic protein for in vitro studies | Functional characterization of desaturation activity and kinetic parameter determination. |
| Fe(II) Salts (e.g., FeSOâ‚„) | Essential Cofactor | Reconstitution of active holoenzyme from purified apo-PolE [5]. |
| L-Isoleucine (L-Ile) | Native Physiological Substrate | Primary substrate for desaturation assays in PolE functional studies [5]. |
| Anaerobic Chamber | Creates oxygen-free environment | Essential for handling and reconstituting oxygen-sensitive Fe²⺠cofactor without oxidation [5]. |
| Oâ‚‚-Saturated Buffer | Oxidant for reaction initiation | Used to start the enzymatic desaturation reaction after anaerobic mixture preparation [5]. |
| Dansyl Chloride (DnsCl) | Derivatization Agent | Enhances detection and analysis of reaction products (L-Ile and 3,4-dehydroIle) by LC-MS [5]. |
| Reductants (Ascorbate/DTT) | External Reducing Agents | Maintains the iron center in its reduced state (Fe²âº) for multiple catalytic turnovers in vitro [5]. |
| Pterin Cofactor | Proposed Electron Transfer Mediator | Required for PolE's oxidative desaturation mechanism, though specific type may require empirical determination [10] [31]. |
| N10-Monodesmethyl Rizatriptan-d3 | N10-Monodesmethyl Rizatriptan-d3|Isotope-Labeled Metabolite | N10-Monodesmethyl Rizatriptan-d3 is a deuterated metabolite for research. For Research Use Only. Not for human or veterinary diagnostic or therapeutic use. |
PolE exemplifies a sophisticated evolutionary solution for optimizing the biosynthesis of complex natural products. By executing a targeted desaturation of L-isoleucine, it overcomes a kinetic bottleneck and provides a streamlined route for the subsequent formation of a highly strained azetidine ring by PolF. Its Fe and pterin-dependent mechanism, combined with its specific substrate recognition, makes it a critical component of the polyoxin biosynthetic pathway. A thorough understanding of PolE's role provides not only fundamental insights into enzymatic catalysis but also opens new avenues for biocatalytic applications. The experimental frameworks and reagent tools detailed in this whitepaper provide a foundation for further research aimed at exploiting this enzyme for the sustainable production of valuable azetidine-containing pharmaceuticals and agrochemicals.
The biosynthesis of azetidine-containing amino acids, such as polyoximic acid (PA) in the antifungal polyoxin pathway, represents a significant biochemical challenge due to the high ring strain involved. Recent research has elucidated a novel pathway where the non-haem iron-dependent enzyme PolF directly catalyzes the transformation of linear amino acids into azetidine derivatives. This whitepaper provides an in-depth technical analysis of the critical roles of Fe(II) and Oâ‚‚ as essential cofactors in this process. We summarize the key experimental findings, detail the methodologies for studying these reactions, and provide actionable guidance for optimizing cofactor conditions to maximize enzymatic activity and azetidine product yield for researchers in enzymology and drug development.
Azetidine is a four-membered nitrogen-containing saturated heterocycle with significant ring strain (approximately 25.4 kcal molâ»Â¹), making it a valuable yet challenging scaffold in medicinal chemistry and drug design [10] [5]. The biosynthesis of azetidine-containing amino acids has long been enigmatic. Prior to the characterization of the PolF enzyme, known biosynthetic mechanisms relied on precursors like S-adenosyl-l-methionine (SAM) or α-ketoglutarate (α-KG), which are metabolically expensive and limit biocatalytic applications [10] [11].
The discovery that PolF, a member of the heme-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily, can directly convert L-isoleucine (L-Ile) and L-valine (L-Val) into azetidine derivatives via a novel iron-dependent mechanism represents a paradigm shift [10] [5]. This technical guide delves into the core cofactor requirements—Fe(II) and O₂—that are fundamental to this enzyme's function. Within the context of a broader thesis on biosynthesis, understanding and optimizing these conditions is paramount for harnessing the full potential of these enzymes in synthesizing pharmaceutically relevant compounds.
The PolF Enzyme and its Cofactor-Dependent Mechanism
PolF is an HDO Enzyme Essential for Azetidine Formation
Genetic knockout experiments in Streptomyces cacaoi, the polyoxin producer, established that PolF is absolutely essential for polyoximic acid biosynthesis. The polF mutant did not produce any measurable polyoxin A (<1% of wild-type), whereas a polE mutant still produced reduced amounts (~10%), highlighting the non-redundant role of PolF [10] [5]. Bioinformatic analysis revealed that PolF belongs to the HDO superfamily, a class of diiron-dependent enzymes that activate molecular oxygen for diverse oxidative reactions [10] [5].
The Catalytic Cycle and Key Intermediates
PolF catalyzes a complex series of transformations on L-Ile and L-Val, including desaturation, hydroxylation, and intramolecular C–N bond formation, to yield the azetidine ring [10]. The mechanism proceeds via a 3,4-desaturated intermediate [5]. Single-turnover experiments with L-Val revealed the formation of 3,4-dehydrovaline (3,4-dhVal) as a major intermediate, which is subsequently converted into the azetidine product 3-methylene-azetidine-2-carboxylic acid (MAA) [10]. This indicates that desaturation precedes cyclization. The reaction is proposed to involve a radical mechanism following an initial, chemically challenging C–H bond cleavage (up to 101 kcal/mol) [10] [5]. Structural and spectroscopic studies identified a μ-peroxo-Fe(III)₂ species as the intermediate directly responsible for this initial H-atom abstraction [10] [5]. The subsequent steps, including the formation of the strained C–N bond, are likely facilitated by radical rearrangements.
The following diagram illustrates the catalytic cycle of PolF, from cofactor loading to product formation.
Experimental Protocols for Cofactor Studies
The following section details the key methodologies used to characterize the Fe(II) and Oâ‚‚ dependence of PolF.
Enzyme Expression and Purification
- Expression System: Heterologous expression of PolF in E. coli [10] [5].
- Purification: Standard protein purification techniques (e.g., affinity chromatography) yield apo-PolF, which is largely iron-free upon initial isolation [10] [5].
- Critical Note: The enzyme is purified in its apo form, necessitating reconstitution with Fe(II) for activity assays.
Diiron Cofactor Reconstitution
- Procedure: Incubate apo-PolF with an excess of Fe(II) (e.g., 3 equivalents) under strict anaerobic conditions [10] [5]. An anaerobic chamber or sealed Schlenk line techniques are required to prevent premature oxidation.
- Removal of Unbound Metal: After incubation, pass the mixture through a desalting column (e.g., PD-10) equilibrated with anaerobic buffer to remove unchelated Fe(II) [10].
- Typical Yield: The reconstituted enzyme typically contains approximately 1.5 equivalents of Fe(II) per monomer, consistent with the weak iron affinity reported for HDO enzymes in the absence of substrate [10] [5].
Standard Activity Assay
This protocol is used to confirm enzymatic activity and its dependence on both Fe(II) and Oâ‚‚.
- Reaction Setup: Prepare the enzyme-substrate mixture anaerobically. Combine reconstituted PolF (e.g., 10-50 µM) with substrate (L-Ile or L-Val, e.g., 1-5 mM) in a sealed vial within an anaerobic chamber [10].
- Initiation: Start the reaction by injecting an Oâ‚‚-saturated buffer [10].
- Reductant Inclusion: Include an external reductant like ascorbate (1-5 mM) or dithiothreitol (DTT) in the assay mixture to recycle the diiron center during multiple turnovers [10] [5].
- Derivatization and Analysis:
Control Experiments
- No Fe(II): Use apo-PolF without reconstitution. Expected result: No product formation [10].
- No Oâ‚‚: Perform the entire assay in an anaerobic environment without adding Oâ‚‚-saturated buffer. Expected result: No product formation [10].
- Heat-Inactivated Enzyme: Use enzyme denatured by boiling. Expected result: No product formation [10].
- Metal Specificity: Test other divalent metals (e.g., Mn(II), Co(II), Ni(II), Cu(II), Zn(II)) during reconstitution. Expected result: Only Fe(II) supports azetidine formation [10].
Quantitative Data on Cofactor Conditions and Enzyme Activity
The following tables summarize key quantitative findings from the functional characterization of PolF.
Table 1: Cofactor Requirements for PolF Activity
| Cofactor / Condition | Concentration / Details | Observed Outcome | Experimental Reference |
|---|---|---|---|
| Fe(II) | 3 eq. for reconstitution | ~1.5 eq. Fe bound per enzyme; Essential for activity | [10] [5] |
| Oâ‚‚ | Oâ‚‚-saturated buffer | Essential for initiating catalysis | [10] [5] |
| External Reductant | Ascorbate or DTT (1-5 mM) | Required for multiple turnovers | [10] |
| Alternative Metals | Mn(II), Co(II), Ni(II), Cu(II), Zn(II) | No azetidine product formed | [10] |
Table 2: Substrate Specificity and Kinetic Intermediates of PolF
| Substrate | Major Product(s) | Mass Change | Key Observation |
|---|---|---|---|
| L-Isoleucine (L-Ile) | Polyoximic Acid (PA) | -4 Da | Native biosynthetic substrate |
| L-Valine (L-Val) | 3-Methylene-azetidine-2-carboxylic acid (MAA) | -4 Da | Major product under multiple turnover |
| L-Valine (Single Turnover) | 3,4-dehydrovaline (3,4-dhVal) | -2 Da | Key desaturated intermediate |
| L-Leucine (L-Leu) | Hydroxylation products (minor desaturation) | +16 Da | No azetidine formed |
| L-Methionine (L-Met) | Hydroxylation products (minor desaturation) | +16 Da | No azetidine formed |
The Scientist's Toolkit: Research Reagent Solutions
The following table lists essential reagents, materials, and instruments required for studying and optimizing PolF-like enzymatic activity.
Table 3: Essential Research Reagents and Materials
| Item | Function / Application | Specific Examples / Notes |
|---|---|---|
| Apo-PolF Enzyme | Catalytic protein for azetidine formation | Heterologously expressed and purified from E. coli [10] [5] |
| Fe(II) Salts | Cofactor for enzyme reconstitution | e.g., Ammonium iron(II) sulfate; handle under anaerobic conditions |
| Anaerobic Chamber | Maintaining oxygen-free environment | Essential for enzyme reconstitution and assay setup to prevent Fe(II) oxidation [10] |
| Desalting Columns | Removal of unbound Fe(II) after reconstitution | e.g., PD-10 Sephadex G-25 columns [10] |
| Oâ‚‚-Saturated Buffer | Oxidant for initiating the catalytic reaction | Prepared by bubbling oxygen through the buffer immediately before use [10] |
| External Reductants | Sustaining multiple enzyme turnovers | Ascorbate or Dithiothreitol (DTT) at 1-5 mM [10] |
| Dansyl Chloride (DnsCl) | Derivatization agent for product detection | Enhances detection of amino acid products in LC-MS [10] |
| LC-MS System | Analysis and quantification of reaction products | Used to identify products based on mass shifts (e.g., -4 Da for azetidine) [10] [5] |
Optimization Strategies and Technical Recommendations
Based on the experimental data, the following recommendations are critical for optimizing cofactor conditions.
- Maintain Strict Anaerobic Handling for Fe(II): The Fe(II) cofactor is highly oxygen-sensitive. All steps involving enzyme reconstitution and reaction setup before Oâ‚‚ addition must be performed in a certified anaerobic chamber or using specialized glassware to preserve the reduced state of the diiron center.
- Utilize a Dual Fe(II) and Reductant System: For maximum catalytic turnover, the reaction mixture should contain both Fe(II)-reconstituted enzyme and a chemical reductant like ascorbate. The Fe(II) loads the active site, while the reductant regenerates the diiron cluster from its oxidized form (Fe(III)) during the catalytic cycle [10].
- Sequential Cofactor Addition is Key: The protocol of first forming the anaerobic PolF–substrate–Fe(II) complex and then initiating the reaction with O₂ is crucial. This sequence mimics the likely natural order of events and ensures the reactive oxygen species is generated in the presence of the substrate, minimizing unproductive decay of the intermediates.
- Monitor for Key Intermediates: Optimization efforts should use LC-MS to track not only the final azetidine product but also the proposed desaturated intermediate (e.g., 3,4-dhVal). An accumulation of this intermediate could indicate a bottleneck in the subsequent cyclization step under suboptimal conditions.
The biosynthesis of azetidine amino acids by PolF is a remarkable feat of enzymatic catalysis, driven decisively by the synergistic roles of Fe(II) and O₂. The Fe(II) diiron center acts as the core reactive unit, while O₂ serves as the driving oxidant, together facilitating difficult C–H activation and radical-mediated ring closure. The experimental protocols and optimization strategies outlined here provide a robust framework for researchers to exploit this novel biosynthesis. Mastering these cofactor conditions is a critical step toward leveraging HDO enzymes like PolF for the sustainable production of high-value, azetidine-containing pharmaceuticals and fine chemicals.
Addressing Substrate Limitations and Stereochemical Constraints
The biosynthesis of azetidine-containing natural products represents a significant frontier in enzymology, particularly given the high ring strain (approximately 25.4 kcal molâ»Â¹) that makes chemical synthesis challenging [5]. For researchers investigating non-haem iron-dependent enzymes in azetidine amino acid biosynthesis, two fundamental challenges persistently emerge: narrow substrate scope and strict stereochemical requirements. These constraints significantly impact the efficiency and applicability of these enzymatic systems for producing diverse azetidine scaffolds for drug discovery.
Recent breakthroughs in elucidating the polyoxin biosynthetic pathway have revealed unprecedented enzymatic strategies to overcome these limitations. This technical guide examines the mechanistic insights from the PolF and PolE enzyme system, providing researchers with experimental frameworks and analytical approaches to address substrate and stereochemical constraints in azetidine biosynthesis.
Current Landscape of Azetidine Biosynthesis
Known Biosynthetic Pathways
Azetidine rings occur in various bioactive compounds, but their biosynthetic formation has remained enigmatic until recently. Previously characterized mechanisms include:
- SAM-dependent cyclization: S-adenosyl-L-methionine (SAM) dependent enzymes catalyze intramolecular nucleophilic cyclization to form azetidine carboxylic acid [5] [11].
- α-KG-dependent oxidative cyclization: α-Ketoglutarate (α-KG) and Fe-dependent oxygenases mediate radical-mediated oxidative C-C bond formation, as observed in okaramine biosynthesis [5].
These established pathways share a significant limitation: dependence on metabolically expensive precursors that restrict their versatility for biocatalytic applications [5] [11]. The recent discovery of an alternative route in the polyoxin pathway provides a fundamentally different approach to azetidine formation.
The Polyoxin Model System
The antifungal nucleoside polyoxin A contains polyoximic acid (PA), an azetidine amino acid derived from L-isoleucine (L-Ile) [5] [11]. Early isotope labeling experiments suggested a distinct biosynthetic mechanism from previously characterized pathways [5]. Within the polyoxin biosynthetic gene cluster, two enzymes—PolE and PolF—were implicated in PA biosynthesis, though their precise functions remained uncharacterized until recently.
Table 1: Key Enzymes in Azetidine Amino Acid Biosynthesis
| Enzyme | Family | Cofactors | Catalytic Function |
|---|---|---|---|
| PolF | HDO (haem-oxygenase-like dimetal oxidase) superfamily | Diiron center (Feâ‚‚) | Transforms L-Ile and L-Val to azetidine derivatives via 3,4-desaturated intermediate |
| PolE | DUF6421 | Fe²⺠and pterin | Catalyzes desaturation of L-Ile, increasing flux through PolF pathway |
| SznF | Multi-domain metalloenzyme | Diiron center | Mediates N-hydroxylation steps in N-nitrosourea pharmacophore of streptozotocin |
Substrate Limitations: Mechanistic Insights and Experimental Approaches
PolF Substrate Specificity and Promiscuity
Gene knockout experiments in Streptomyces cacaoi established that PolF is absolutely essential for PA biosynthesis, with the polF mutant producing no measurable polyoxin A (<1% of wild-type) [5]. In vitro characterization of PolF revealed both expected substrate preferences and unexpected promiscuity:
Table 2: PolF Substrate Specificity Profile
| Substrate | Major Product(s) | Relative Activity | Notes |
|---|---|---|---|
| L-isoleucine | Polyoximic acid (PA) | +++ | Native pathway substrate |
| L-valine | 3-methylene-azetidine-2-carboxylic acid (MAA) | +++ | Efficient azetidine formation |
| L-leucine | Hydroxylation products (minor desaturation) | + | No azetidine formation |
| L-methionine | Hydroxylation products (minor desaturation) | + | No azetidine formation |
| Other proteogenic amino acids | No detectable products | - | High selectivity for medium-size aliphatic amino acids |
Experimental data indicates that PolF reacts selectively with medium-size aliphatic amino acids, with the β-methyl group being critical for azetidine formation [5]. The enzyme's ability to process both L-Ile and L-Val to azetidine products demonstrates a degree of substrate flexibility, while its failure to produce azetidine rings from L-Leu or L-Met reveals specific steric constraints.
Experimental Protocol: Substrate Screening
Objective: Determine enzyme activity across potential substrate analogs.
Methodology:
- Express and purify PolF from E. coli (apo form)
- Reconstitute with 3 equivalents Fe(II) under anaerobic conditions
- Remove excess Fe(II) using desalting column
- Prepare enzyme-substrate mixtures anaerobically
- Initiate reactions with Oâ‚‚-saturated buffer
- Derivatize products with dansyl chloride (DnsCl)
- Analyze by LC-MS monitoring for -4 Da mass shift (characteristic of azetidine formation)
Critical Controls:
- Reactions without Fe(II)
- Reactions without Oâ‚‚
- Heat-inactivated PolF
- Assays with alternative transition metals
Interpretation: Authentic azetidine formation requires all reaction components, with only Fe(II) supporting catalysis [5]. The dependence on external reductants (ascorbate or dithiothreitol) confirms the need for re-reduction of the diiron center during multiple turnovers.
Stereochemical Constraints: Analysis and Engineering Strategies
Stereochemical Flexibility of PolF
The stereochemical requirements of PolF were investigated using L-Ile stereoisomers, revealing unexpected flexibility:
- L-Ile: Efficient conversion to PA (native substrate)
- L-allo-Ile: Reduced but detectable azetidine formation
- D-Ile: Reduced but detectable azetidine formation
- D-allo-Ile: Reduced but detectable azetidine formation
These findings indicate that while the stereochemistries at C2 and C3 impact catalytic efficiency, they are not absolute determinants of substrate recognition [5]. This moderate stereochemical flexibility expands the potential substrate range for biocatalytic applications.
Strategic Approach to Stereochemical Constraints
The structural basis for this stereochemical permissibility can be leveraged through several strategic approaches:
- Enzyme Engineering: Modify active site residues to accommodate diverse stereochemical configurations
- Cofactor Manipulation: Alter the diiron cluster coordination to modulate stereoselectivity
- Reaction Optimization: Adjust reaction conditions to favor desired stereochemical outcomes
Mechanistic Framework for C-N Bond Formation
Reaction Intermediates and Pathway Branching
Single-turnover experiments with L-Val as substrate revealed that PolF catalyzes three distinct reactions—desaturation, hydroxylation, and C-N bond formation—on a single substrate [5]. Under multiple turnover conditions, the major product is the azetidine derivative MAA, while single-turnover studies identified key intermediates:
- 3,4-dehydrovaline (3,4-dh-Val): Major initial product (0.23 minâ»Â¹ formation rate)
- 3-dimethylaziridine-2-carboxylic acid (Azi): Intermediate with C-N bond (0.15 minâ»Â¹)
- 4-hydroxyvaline (4-OH-Val): Hydroxylation product (0.14 minâ»Â¹)
- 3-hydroxyvaline (3-OH-Val): Hydroxylation product (0.036 minâ»Â¹)
The identification of 3,4-dh-Val as the major initial product and its quantitative conversion to MAA establishes it as a key intermediate in azetidine formation [5].
Experimental Protocol: Intermediate Trapping
Objective: Characterize reaction intermediates to establish catalytic mechanism.
Methodology:
- Perform PolF assays under single-turnover conditions (2 equivalents Fe²âº, no external reductant)
- Monitor time-dependent formation of products using LC-MS
- Compare retention times and mass spectra to authentic standards where available
- Isolate intermediates for structural characterization by NMR
- Use substrate analogs to trap specific intermediates
Key Findings: The desaturated intermediate (3,4-dh-Val) is quantitatively converted to the azetidine product, establishing it as a key intermediate in the pathway [5]. The detection of Azi demonstrates PolF's inherent capacity for C-N bond formation independent of the azetidine ring.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Azetidine Biosynthesis Research
| Reagent/Category | Function/Application | Specific Examples | Experimental Notes |
|---|---|---|---|
| Enzyme Expression | Recombinant protein production | PolF, PolE in E. coli | PolF initially isolated in apo form; requires Fe reconstitution |
| Cofactors | Essential catalytic components | Fe(II), pterin (for PolE), Oâ‚‚ | Requires anaerobic handling for Fe(II) reconstitution |
| Activity Assay Reagents | Reaction monitoring and analysis | Dansyl chloride (DnsCl), ascorbate, dithiothreitol | DnsCl for product derivatization; ascorbate/DTT as external reductants |
| Analytical Standards | Product identification and quantification | Polyoximic acid (from polyoxin A hydrolysis), 3,4-dehydrovaline | Critical for LC-MS method development and validation |
| Chromatography | Product separation and analysis | Reverse-phase LC columns, desalting columns | Desalting columns for excess Fe(II) removal after reconstitution |
| Spectroscopy | Structural and mechanistic studies | X-ray crystallography, Mössbauer spectroscopy | Crystal structures of PolF with substrates reveal binding mode |
Integrated Experimental Workflow
The discovery that PolF utilizes a μ-peroxo-Fe(III)â‚‚ species to catalyze chemically challenging C-H activation (up to 101 kcal molâ»Â¹) provides a new paradigm for azetidine ring formation [5]. The enzyme's ability to process multiple substrates with moderate stereoselectivity, coupled with its capacity to catalyze both desaturation and C-N bond formation, offers unprecedented opportunities for biocatalytic applications.
Future research directions should focus on:
- Engineering PolF for expanded substrate range while maintaining azetidine-forming activity
- Harnessing the stereochemical flexibility to produce diverse azetidine scaffolds
- Integrating PolF/PolE system with other biocatalytic modules for synthesis of complex azetidine-containing molecules
- Exploiting the mechanistic insights to design biomimetic catalytic systems
The experimental frameworks and technical approaches detailed in this guide provide a foundation for addressing the persistent challenges of substrate limitations and stereochemical constraints in azetidine biosynthesis, potentially unlocking new avenues for pharmaceutical development targeting this structurally distinctive class of amino acids.
Improving Enzyme Efficiency and Stability for Industrial Applications
The pursuit of efficient and stable biocatalysts is a central goal in industrial enzymology, particularly for complex chemical transformations. Non-haem iron-dependent enzymes have emerged as particularly promising targets due to their ability to catalyze chemically challenging reactions under mild conditions. Recent research has illuminated the remarkable capabilities of these enzymes, especially in the biosynthesis of azetidine-containing amino acids—high-value strained heterocycles with significant pharmaceutical potential [32] [33]. The discovery that non-haem iron enzymes can construct these coveted four-membered rings biologically represents a transformative advance with profound implications for green chemistry and drug development [33].
Industrial applications of enzymes frequently encounter significant hurdles, including insufficient catalytic efficiency, operational instability, and limited functional expression in heterologous systems. These challenges are particularly pronounced for non-haem iron enzymes, which often exhibit structural fragility despite their catalytic versatility [34]. This technical guide examines recent breakthroughs in understanding, measuring, and enhancing the performance of non-haem iron enzymes, with specific emphasis on their application in azetidine biosynthesis. By integrating foundational mechanistic insights with advanced engineering strategies, we provide a comprehensive framework for transforming these promising biocatalysts into robust industrial tools.
Azetidine Biosynthesis: A Case Study in Non-Haem Iron Enzyme Function
Mechanistic Insights from the Polyoxin Pathway
The biosynthetic pathway for the antifungal nucleoside polyoxin A has recently yielded crucial insights into azetidine formation by non-haem iron enzymes. Within this pathway, two key iron-dependent enzymes—PolF and PolE—orchestrate the conversion of standard amino acids into azetidine-containing derivatives [32] [10].
PolF represents a novel member of the haem-oxygenase-like dimetal oxidase/oxygenase (HDO) superfamily. Remarkably, this enzyme alone can transform L-isoleucine (L-Ile) and L-valine into their corresponding azetidine derivatives via a 3,4-desaturated intermediate [32]. Mechanistic and structural studies indicate that PolF utilizes a μ-peroxo-Fe(III)₂ intermediate to cleave unactivated C-H bonds (with bond dissociation energies up to 101 kcal/mol), with subsequent C-N bond formation proceeding through radical mechanisms [10].
PolE, a member of the DUF6421 family, functions as an iron and pterin-dependent oxidase that catalyzes the desaturation of L-Ile, thereby increasing the flux through the PolF-catalyzed pathway [32] [10]. This synergistic relationship demonstrates how multiple non-haem iron enzymes can cooperate to enhance overall pathway efficiency.
Table 1: Key Non-Haem Iron Enzymes in Azetidine Biosynthesis
| Enzyme | Structural Family | Cofactors | Catalytic Function | Azetidine Products |
|---|---|---|---|---|
| PolF | HDO superfamily | Diiron center | Azetidine ring formation from L-Ile/L-Val | Polyoximic acid, 3-methylene-azetidine-2-carboxylic acid |
| PolE | DUF6421 family | Fe²âº, pterin | Substrate desaturation | 3,4-dehydroisoleucine (intermediate) |
| AzeJ | Class I methyltransferase fold | None (SAM substrate) | SAM cyclization to azetidine-2-carboxylic acid | Azetidine-2-carboxylic acid |
Comparative Azetidine Biosynthesis Mechanisms
Beyond the PolF/PolE system, alternative enzymatic routes to azetidine rings have been characterized, providing valuable comparative insights. AzeJ and VioH represent SAM-dependent azetidine synthases that catalyze intramolecular 4-exo-tet cyclization of S-adenosylmethionine (SAM) to form azetidine-2-carboxylic acid [35]. Structural analyses reveal that these enzymes position the SAM substrate in a kinked conformation that facilitates nucleophilic attack of the nitrogen on the Cγ-carbon, with the positive charge of the sulfonium group migrating to stabilize the newly formed C-N bond [35].
The existence of distinct azetidine-forming mechanisms—radical-based non-haem iron chemistry versus SAM cyclization—highlights the functional diversity of enzymes capable of constructing these strained rings and provides multiple engineering starting points for industrial applications.
Quantitative Assessment of Enzyme Performance
Robust quantification of enzyme performance parameters is essential for evaluating improvement strategies. The following metrics provide critical benchmarks for assessing non-haem iron enzymes in industrial contexts.
Table 2: Key Performance Metrics for Non-Haem Iron Enzymes
| Performance Metric | Definition | Measurement Methods | Industrial Significance |
|---|---|---|---|
| Total Turnover Number (TTN) | Moles of product per mole of enzyme before inactivation | LC-MS of timed reactions | Determines enzyme lifetime and process economics |
| Catalytic Efficiency (kâ‚ₜₜ/Kₘ) | Specificity constant measuring catalytic proficiency | Michaelis-Menten kinetics | Predicts performance under substrate-limiting conditions |
| Thermal Stability (Tâ‚…â‚€) | Temperature at which 50% activity is lost after fixed time | Thermofluor assays, activity measurements after heating | Determines process temperature constraints |
| Solubility/Expression Yield | mg of soluble, active enzyme per liter of culture | SDS-PAGE, activity assays | Impacts production costs and scalability |
| Cofactor Affinity (Kₘ) | Michaelis constant for Fe²âº, αKG, or other cofactors | Activity with varying cofactor concentrations | Predicts sensitivity to cofactor depletion |
Recent studies illustrate the application of these metrics. For example, the Fe(II)/αKG enzyme tP4H demonstrated a TTN of approximately 5 for its non-native carboxylate substrate 1, roughly 130-fold lower than its TTN for the native amino acid substrate [34]. This performance gap highlights the substantial improvement potential for non-native reactions. Additionally, instability manifestations—including rapid activity loss and poor expression—were directly quantified during tP4H characterization [34].
Experimental Protocols for Characterizing Non-Haem Iron Enzymes
Enzyme Activity Assays Under Anaerobic Conditions
Purpose: To measure the catalytic activity of oxygen-sensitive non-haem iron enzymes while maintaining the reduced state of the iron center.
Methodology:
- Prepare apo-enzyme by purifying target protein (e.g., PolF) from E. coli and removing bound metals via chelating agents or dialysis [10].
- Reconstitute active enzyme by incubating apo-enzyme with excess Fe(II) (typically 2-3 equivalents) under anaerobic conditions (oxygen-free glove box or sealed Schlenk line) [10].
- Remove unbound iron using desalting columns equilibrated with anaerobic buffer.
- Determine iron content via colorimetric assays or atomic absorption spectroscopy to verify proper reconstitution (target: ~2 Fe per enzyme for diiron centers) [10].
- Prepare substrate solutions in oxygen-free buffers and introduce to enzyme anaerobically.
- Initiate reactions by adding Oâ‚‚-saturated buffer to achieve desired final oxygen concentration.
- Monitor reaction progress via timed aliquots, analyzing products by LC-MS, often after derivatization with reagents like dansyl chloride for enhanced detection [10].
- Include controls without enzyme, without Fe(II), without Oâ‚‚, and with heat-inactivated enzyme to validate reaction specificity [10].
Structural Characterization of Enzyme-Substrate Complexes
Purpose: To visualize atomic-level interactions between non-haem iron enzymes and their substrates/products for rational engineering.
Methodology:
- Express and purify target enzyme (e.g., AzeJ, VioH) with affinity tags (His-tag) followed by size-exclusion chromatography [35].
- Conduct crystallization trials using robotic screening systems with commercial sparse matrix screens.
- Optimize initial hits by varying pH, precipitant concentration, and temperature.
- Soak crystals with substrates (SAM) or analogs (SAH) or co-crystallize with ligands [35].
- Flash-cool crystals in liquid nitrogen with appropriate cryoprotectants.
- Collect X-ray diffraction data at synchrotron facilities.
- Solve structures by molecular replacement using homologous structures (e.g., DDMT-N1-MT for AzeJ) [35].
- Analyze active site architecture, substrate positioning, and conformational changes between liganded and unliganded states.
Computational Stabilization Using ProteinMPNN
Purpose: To enhance enzyme stability and expression while maintaining catalytic function through deep learning-based sequence redesign.
Methodology:
- Obtain or generate a high-quality protein structure (experimental or AlphaFold2 model) of target enzyme [34].
- Identify and fix catalytically essential residues (Fe(II) coordination, substrate binding, cofactor interaction) to preserve function [34].
- Optionally fix additional residues based on conservation (multiple sequence alignment) or spatial proximity (within 10Ã… of active site) [34].
- Use ProteinMPNN to generate redesigned sequences with optimized stability metrics while constraining fixed positions [34].
- Select designed variants based on computational metrics (top-ranked Cα-RMSD to input structure) [34].
- Synthesize and clone selected variants for experimental testing.
- Express and purify stabilized variants, comparing expression yield, solubility, and thermal stability to wild-type enzyme.
- Validate retained catalytic function through activity assays with native and non-native substrates.
Strategic Approaches to Enhancement
Active Site Engineering with Functional Preservation
Engineering non-haem iron enzymes for improved performance requires balancing stability enhancements with catalytic function preservation. Critical considerations include:
- Identify Essential Residues: Based on structural and mechanistic studies, residues involved in iron coordination (typically His, Asp, Glu residues in 2-His-1-carboxylate facial triad), substrate positioning, and cofactor binding must be conserved [34] [36].
- Define Second-Sphere Interactions: Residues beyond direct coordination but contributing to active site architecture (e.g., Phe134 in AzeJ, which stabilizes reaction intermediates through cation-Ï€ interactions) should typically be preserved [35].
- Leverage Natural Diversity: Analyze homologous enzymes with different stability profiles but similar functions to identify beneficial substitutions outside conserved regions.
Cofactor Engineering and Cellular Redox Management
Non-haem iron enzymes frequently require maintaining iron in the reduced Fe(II) state and managing reactive oxygen species generated during catalysis:
- Metallochaperone Co-expression: Express iron-binding proteins (e.g., IscA, SufA) to enhance proper metallocenter assembly [10].
- Redox Buffer Systems: Include low-molecular-weight reductants (ascorbate, DTT) in reaction buffers to maintain iron in reduced state and support multiple turnover [10].
- Cellular Redox Engineering: For whole-cell biocatalysis, engineer host strains with enhanced reducing capacity (e.g., NADPH regeneration) or oxidative stress resistance.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Non-Haem Iron Enzyme Studies
| Reagent Category | Specific Examples | Function/Application | Technical Considerations |
|---|---|---|---|
| Expression Systems | E. coli BL21(DE3), BAP1 | Heterologous enzyme production | BAP1 strain expresses membrane-bound phosphatase to enhance metal uptake |
| Purification Tags | His-tag, Strep-tag | Affinity purification | His-tag may interfere with metal content; consider removal after purification |
| Anaerobic Workstations | Coy Laboratory Products, Belle Technology | Maintain oxygen-sensitive enzymes and reactions | Typically maintain <1 ppm Oâ‚‚ with Hâ‚‚/Nâ‚‚ mix and palladium catalysts |
| Iron Cofactors | FeSOâ‚„, FeClâ‚‚, Fe(NHâ‚„)â‚‚(SOâ‚„)â‚‚ | Enzyme reconstitution | Prepare fresh solutions in acidic anaerobic buffers to prevent oxidation |
| Structural Biology Reagents | SAM, SAH, substrate analogs | Crystallization and mechanistic studies | SAH serves as stable SAM analog for structural studies |
| Analytical Standards | Polyoximic acid, azetidine-2-carboxylic acid | LC-MS quantification | Isolate from natural sources or synthesize chemically for calibration |
Visualization of Engineering Workflows
Diagram 1: A structured workflow for computationally-guided enzyme stabilization, integrating deep learning-based redesign with experimental validation.
Diagram 2: The coordinated biosynthetic pathway for azetidine formation showing the sequential actions of PolE and PolF on L-isoleucine.
The strategic enhancement of non-haem iron enzyme efficiency and stability represents a cornerstone of next-generation industrial biocatalysis. Recent elucidation of azetidine biosynthesis pathways has not only revealed novel enzymatic mechanisms but also provided exemplary systems for developing and validating improvement strategies. The integration of computational design tools like ProteinMPNN with traditional enzyme engineering approaches creates powerful synergies for overcoming the intrinsic limitations of these catalytically versatile but often fragile enzymes.
As structural and mechanistic understanding of non-haem iron enzymes continues to advance, particularly through techniques such as X-ray crystallography and spectroscopic characterization, rational engineering efforts will become increasingly precise and effective. The expanding repertoire of documented azetidine-forming mechanisms provides multiple evolutionary starting points for engineering campaigns aimed at specific industrial applications. By applying the systematic approaches outlined in this guide—combining rigorous quantification, strategic residue selection, and holistic cellular engineering—researchers can transform these promising biocatalysts into robust industrial workhorses capable of enabling sustainable manufacturing processes for high-value chemical targets.
Validating the Pathway and Comparative Analysis with Established Mechanisms
Within the broader context of research on the biosynthesis of azetidine amino acids by non-haem iron enzymes, the critical challenge lies in experimentally characterizing the transient metal centers and reactive species that catalyze these transformations. The biosynthesis of polyoximic acid, the azetidine-containing amino acid found in the antifungal polyoxin A, represents a paradigm shift in our understanding of enzymatic azetidine formation [5]. Early isotope-labelling experiments suggested this moiety was derived from L-isoleucine, but the enzymatic mechanism remained enigmatic for decades [5]. Recent breakthroughs have identified PolF, a member of the haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily, as the enzyme responsible for the remarkable transformation of L-isoleucine into an azetidine product via a 3,4-desaturated intermediate [5] [6]. This technical guide provides a comprehensive framework for the spectroscopic validation of the diiron center and key intermediates in this and related biosynthetic pathways, with particular emphasis on methodologies relevant to the PolF system.
Research Reagent Solutions for Spectroscopic Characterization
The following table details essential reagents and materials required for experiments aimed at characterizing non-haem diiron centers and their intermediates.
Table 1: Key Research Reagents and Their Applications
| Reagent / Material | Function in Spectroscopic Analysis |
|---|---|
| PolF Enzyme (HDO Superfamily) | Primary catalyst for azetidine formation; contains the dimetal center for spectroscopic interrogation [5]. |
| L-Isoleucine (L-Ile) / L-Valine | Native substrates for PolF; enable trapping of intermediates during catalytic cycle [5]. |
| 57Fe-Enriched Samples | Mössbauer spectroscopy; provides detailed information on oxidation states, spin states, and electronic environment of iron centers [37]. |
| Chemical Reductants (Ascorbate/DTT) | Maintains diiron center in reduced Fe(II)2 state for O2 activation during multiple turnover experiments [5]. |
| O2-Saturated Buffers | Initiation of catalytic turnover for trapping reactive oxygen intermediates (e.g., μ-peroxo species) [5]. |
| Deuterated Solvents | Nuclear Resonance Vibrational Spectroscopy (NRVS); minimizes interference from proton vibrations [37]. |
| Cryogenic Solutions | Stabilization of metastable intermediates for EPR, Mössbauer, and rRaman spectroscopy [38] [39]. |
| Authentic Standards (e.g., PA, 3,4-dh-Val) | LC-MS calibration for identifying and quantifying enzymatic reaction products [5]. |
Experimental Workflow for Intermediate Trapping and Analysis
The following diagram outlines the core experimental workflow for generating, trapping, and characterizing reactive intermediates in non-haem diiron enzyme systems like PolF.
Figure 1. Experimental workflow for trapping and analyzing diiron enzyme intermediates. The process begins with preparing the metal-free enzyme (Apo-Enzyme), followed by anaerobic incorporation of iron and substrate binding. The reaction is initiated by oxygen introduction, and intermediates are trapped at precise time points using rapid freeze-quench techniques for parallel analysis with multiple spectroscopic methods.
Detailed Methodologies for Key Experiments
Enzyme Preparation and Reconstitution of the Diiron Center
Protocol 1: Expression, Purification, and Metallic Reconstitution of PolF
- Expression and Purification: Heterologously express PolF in E. coli and purify using affinity and size-exclusion chromatography. The enzyme is initially isolated in a largely apo (metal-free) form [5].
- Anaerobic Reconstitution: Perform all subsequent steps in an anaerobic glove box (<1 ppm O2). Incubate apo-PolF with a 1.5-3 molar equivalents of Fe(II) salt (e.g., Fe(NHâ‚„)â‚‚(SOâ‚„)â‚‚) [5].
- Removal of Unbound Metal: Pass the reconstitution mixture through a desalting column (e.g., PD-10) equilibrated with anaerobic buffer to remove excess, unbound Fe(II). Metal analysis typically confirms incorporation of approximately 1.5-2.0 equivalents of Fe per enzyme [5].
- Substrate Binding: Incubate the reconstituted holo-enzyme with substrate (L-Ile or L-Val) under anaerobic conditions. Substrate binding often enhances the affinity and stability of the diiron cluster [5].
Trapping the μ-Peroxo-Fe(III)₂ Intermediate
Protocol 2: Single-Turnover Reaction for Intermediate Characterization
- Sample Preparation: Mix the substrate-bound, Fe(II)-reconstituted PolF in an anaerobic environment. Use a stoichiometric amount of Fe (2 equivalents) relative to the enzyme, without external reductants, to allow only a single catalytic turnover [5].
- Rapid Mixing and Freeze-Quench: Rapidly mix the enzyme-substrate complex with an equal volume of Oâ‚‚-saturated buffer using a specialized stopped-flow or rapid-quench instrument.
- Cryogenic Trapping: At a specific, empirically determined time (e.g., 10-500 ms), eject the reaction mixture into an isopentane bath cooled to near -140°C, or into a specialized EPR tube submerged in liquid nitrogen. This halts the reaction and traps the intermediate(s).
- Spectroscopic Analysis: Analyze the frozen sample directly using low-temperature spectroscopic techniques. This single-turnover approach with PolF using L-Val as a substrate has successfully allowed for the accumulation of the μ-peroxo-Fe(III)₂ species and other intermediates like 3,4-dehydrovaline (3,4-dh-Val) for characterization [5].
Spectroscopic Signatures and Data Interpretation
Key Spectroscopic Techniques and Parameters
The following table summarizes the core spectroscopic techniques and the key parameters they provide for characterizing diiron centers.
Table 2: Spectroscopic Techniques for Diiron Center Analysis
| Technique | Information Provided | Key Parameters Measured | Example Signature in PolF/Models |
|---|---|---|---|
| Mössbauer Spectroscopy | Oxidation & spin state of each Fe; electron delocalization [37]. | Isomer shift (δ, mm/s); Quadrupole splitting (ΔEQ, mm/s). | A μ-peroxo-Fe(III)₂ site shows two similar quadrupole doublets indicative of antiferromagnetically coupled high-spin Fe(III) ions [5] [39]. |
| EPR Spectroscopy | Presence of paramagnetic species (radicals, Fe centers); spin state of cluster [38] [37]. | g-values. | A resting Fe(II)2 center is EPR-silent. A μ-peroxo-Fe(III)₂ intermediate is typically anti-ferromagnetically coupled and also EPR-silent. Radical intermediates may be detectable [5] [39]. |
| Resonance Raman (rRaman) | Identity of Fe-ligand vibrations (Fe-O, Fe-S); peroxide binding mode [38] [39]. | Vibrational frequencies (cmâ»Â¹). | An Fe(III)-OCl model complex showed a characteristic Fe–O vibration; similar Fe–O and O–O stretches are expected for a μ-1,2-peroxo bridge [38]. |
| NRVS | Low-energy vibrations involving Fe motion; complete vibrational density of states [37]. | Vibrational frequencies and intensities. | In Fe-Te clusters, NRVS directly probes the low-energy "PKS" vibration critical for understanding electron delocalization and spin states [37]. |
| UV-Vis Absorption | Electronic transitions; charge-transfer bands indicative of specific intermediates [38]. | Wavelength (λmax, nm); Extinction coefficient (ε, Mâ»Â¹cmâ»Â¹). | A biomimetic Fe(III)-OCl complex displayed a distinct absorption band at ~480 nm [38]. |
Quantitative Spectroscopic Data from Model Complexes
Table 3: Experimental Spectroscopic Data from Relevant Iron Complexes
| Complex / Intermediate | Technique | Experimental Data | Interpretation |
|---|---|---|---|
| [Feᴵᴵᴵ(OCl)(MeN4Py)]²⺠[38] | UV-Vis | λmax = 480 nm (ε > 500 Mâ»Â¹cmâ»Â¹) | Ligand-to-metal charge transfer band characteristic of Fe(III)-hypohalite unit. |
| EPR | g = 2.26, 2.15, 1.97 | Signals characteristic of a low-spin iron(III) complex. | |
| Diiron(III)-μ-oxo with Thiolate Ligands [39] | Mössbauer | δ = 0.50-0.55 mm/s; ΔEQ = 0.90-1.70 mm/s | Parameters consistent with antiferromagnetically coupled high-spin Fe(III) sites. |
| [Feâ‚‚Teâ‚‚]+ Cluster (S=3/2) [37] | Mössbauer | Single quadrupole doublet | Evidence of a fully delocalized, mixed-valent Fe²·âµâº-Fe²·âµâº (Robin-Day Class III) electronic structure. |
| EPR | Signals at g ~ 4.3, 3.6 | Signature of an intermediate S = 3/2 spin state. |
The sophisticated application of a multi-technique spectroscopic toolkit is indispensable for demystifying the structure and function of diiron centers in enzymes like PolF. The guided workflows and reference data provided here serve as a blueprint for researchers to rigorously validate the existence of proposed intermediates, such as the μ-peroxo-Fe(III)₂ species in azetidine biosynthesis. Mastering these techniques paves the way for a deeper mechanistic understanding of non-heme iron enzymes, facilitating their potential application in biocatalysis and rational drug design.
Azetidine amino acids are valuable building blocks in medicinal chemistry due to the high ring strain and unique bioactivity imparted by their four-membered nitrogen-containing heterocycle [5]. Within natural product biosynthesis, such as the antifungal polyoxin A and various bacterial peptides, nature has evolved distinct enzymatic strategies to overcome the significant energy barrier (approximately 25.4 kcal molâ»Â¹) required for azetidine ring formation [5] [40]. Recent research has elucidated three primary enzymatic pathways responsible for this transformation, each employing different metallo-cofactors and catalytic mechanisms. This review provides a comparative analysis of the Haem-oxygenase-like Dimetal Oxidase (HDO), S-adenosyl-L-methionine (SAM)-dependent, and α-ketoglutarate (αKG)-dependent pathways, focusing on their mechanistic principles, structural features, and biotechnological applications for researchers and drug development professionals.
The table below summarizes the core characteristics of the three principal enzymatic pathways for azetidine biosynthesis.
Table 1: Comparative Overview of Azetidine Biosynthetic Pathways
| Feature | HDO Pathway | SAM-Dependent Pathway | αKG-Dependent Pathway |
|---|---|---|---|
| Representative Enzyme | PolF (Polyoxin biosynthesis) [5] | AzeJ, VioH (Azetidomonamides, Vioprolides) [40] | TqaL (Fungal alkaloids) [41] |
| Primary Cofactors | Diiron center (Fe₂) [5] | S-adenosylmethionine (SAM) [40] | Fe(II), α-ketoglutarate (αKG) [41] |
| Core Substrate | L-Isoleucine, L-Valine [5] | S-adenosylmethionine (SAM) [40] | Proteinogenic amino acid precursors (varies) [41] |
| Key Reaction Type | Radical-based desaturation & cyclization [5] | Intramolecular nucleophilic substitution (SN2) [40] | Radical-mediated C–N bond formation [41] |
| Metal Center Role | O2 activation, H-atom abstraction via μ-peroxo-Fe(III)2 [5] | Electrophilic activation of sulfonium center [40] | O2 activation, H-atom abstraction via Fe(IV)=O [41] |
| Byproducts | H2O [5] | 5'-Methylthioadenosine (MTA) [40] | Succinate, CO2 [41] |
Detailed Mechanistic Analysis
The HDO Pathway: PolF and Radical-Mediated Cyclization
The HDO pathway, exemplified by PolF in polyoxin biosynthesis, represents a recently characterized route that directly converts branched-chain amino acids like L-Ile into azetidine products (e.g., polyoximic acid) [5]. This enzyme is a member of the haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily.
- Catalytic Cycle and Key Intermediate: PolF utilizes a diiron (Fe2) cluster to activate molecular oxygen. Mechanistic studies suggest the formation of a μ-peroxo-Fe(III)2 intermediate, which is directly responsible for the challenging cleavage of an unactivated C–H bond in the substrate [5]. Subsequent steps, including a critical C–N bond formation to create the four-membered ring, are proposed to proceed through radical mechanisms [5].
- Multi-step Transformation: Under single-turnover conditions, PolF catalyzes three distinct reactions on a single substrate: desaturation, hydroxylation, and C–N bond formation [5]. The desaturated intermediate (3,4-dehydrovaline from L-Val) is a key precursor quantitatively converted to the final azetidine product [5].
- Auxiliary Enzyme (PolE): The pathway efficiency is enhanced by PolE, an Fe- and pterin-dependent oxidase that catalyzes the desaturation of L-Ile, thereby increasing the substrate flux through PolF [5].
The SAM-Dependent Pathway: AZE Synthases and Intramolecular Cyclization
SAM-dependent azetidine synthases, such as AzeJ and VioH, catalyze the formation of azetidine-2-carboxylic acid (AZE) through a direct intramolecular cyclization of SAM, concurrently releasing 5'-methylthioadenosine (MTA) [40] [42].
- Structural Basis and Catalysis: These enzymes typically adopt a Rossmann fold and belong to the class I methyltransferase family [40] [43]. The cyclization is facilitated by a unique "kinked" conformation of the SAM substrate within the active site, which positions the nucleophilic nitrogen atom near the electrophilic Cγ-carbon of the sulfonium group [40].
- Transition State Stabilization: The reaction proceeds via an SN2-type nucleophilic attack. Crystal structures of AzeJ in complex with SAH (S-adenosylhomocysteine) and the products (MTA and AZE) reveal that the transition state and product are stabilized by specific protein interactions. These include hydrogen bonds with residues like Tyr175 and Asn139, and crucial cation-Ï€ interactions between the protonated azetidine nitrogen and Phe134, which help stabilize the positive charge that migrates from the sulfonium during cyclization [40].
The α-Ketoglutarate-Dependent Pathway
αKG-dependent oxygenases constitute a large superfamily of non-haem iron enzymes that catalyze diverse oxidative reactions, including the formation of strained heterocycles like aziridines and azetidines [41] [44].
- Generic Reaction Cycle: These enzymes use Fe(II) and αKG as essential cofactors. The canonical mechanism involves the decarboxylation of αKG, consuming one oxygen atom to produce succinate and CO2, while generating a highly reactive Fe(IV)=O (ferryl-oxo) species [41] [44].
- Azetidine Formation: The powerful Fe(IV)=O intermediate abstracts a hydrogen atom from the substrate, generating a substrate radical. This radical then undergoes rearrangement and intramolecular C–N bond formation to yield the azetidine ring. The enzyme TqaL is a fungal Fe(II)/αKG-dependent oxygenase that catalyzes such a unique C–N bond formation for azetidine synthesis in alkaloid pathways [41].
Experimental Protocols for Key Enzymes
In vitro Assay for HDO Enzyme PolF
Objective: To reconstitute the activity of recombinant PolF and detect the formation of azetidine products from L-Ile or L-Val [5].
Protein Purification and Reconstitution:
- Heterologously express and purify PolF from E. coli (typically as an apo-protein).
- Under anaerobic conditions (e.g., in an glove box), incubate apo-PolF with a 3-fold molar excess of Fe(II) (e.g., (NHâ‚„)â‚‚Fe(SOâ‚„)â‚‚).
- Remove unbound iron using a desalting column (e.g., PD-10) pre-equilibrated with an anaerobic buffer.
Enzyme Assay Setup:
- Prepare the reaction mixture anaerobically containing: 50 mM HEPES buffer (pH 7.5), 2-5 µM Fe-reconstituted PolF, 1-2 mM substrate (L-Ile or L-Val), and 2-5 mM external reductant (e.g., ascorbate or dithiothreitol).
- Initiate the reaction by adding an Oâ‚‚-saturated buffer to achieve final dissolved oxygen concentration.
Product Analysis:
- Quench the reaction after a specific time (e.g., 30-60 min).
- Derivatize amino acids in the reaction mixture with dansyl chloride (DnsCl).
- Analyze the derivatized products by Liquid Chromatography-Mass Spectrometry (LC-MS). Identify polyoximic acid (from L-Ile) or 3-methylene-azetidine-2-carboxylic acid (MAA, from L-Val) by comparing their molecular weights (-4 Da vs. substrate) and retention times to authentic standards [5].
Structural Analysis of SAM-Dependent AZE Synthase
Objective: To determine the high-resolution structure of an AZE synthase (e.g., AzeJ) in complex with its substrate (SAM) or product (SAH/AZE) to elucidate the catalytic mechanism [40].
Protein Crystallization:
- Purify recombinant AzeJ to homogeneity via Ni²âº-affinity and size-exclusion chromatography.
- Crystallize the protein using the sitting-drop vapor-diffusion method. Co-crystallize with 1-2 mM SAM or SAH, as the ligand-free protein may be recalcitrant to crystallization.
Data Collection and Structure Determination:
- Flash-cool crystals in liquid nitrogen using a suitable cryoprotectant.
- Collect X-ray diffraction data at a synchrotron source.
- Solve the structure by molecular replacement using a related Rossmann-fold methyltransferase as a search model.
- Iteratively build and refine the atomic model. The Fo-Fc electron density map will clearly show the bound ligand (SAH or MTA/AZE).
Mechanistic Insights:
- Analyze the active site geometry to identify the kinked conformation of SAH/SAM.
- Map the hydrogen-bonding network and cation-Ï€ interactions (e.g., with Phe134) that stabilize the transition state and the protonated AZE product [40].
Pathway Visualization and Research Toolkit
Comparative Catalytic Mechanisms
The following diagram illustrates the core catalytic cycles and key intermediates for the three azetidine-forming pathways.
Essential Research Reagents and Materials
Table 2: Key Reagent Solutions for Azetidine Biosynthesis Research
| Reagent/Material | Specific Example | Function in Research |
|---|---|---|
| Cloning & Expression | E. coli BL21(DE3) cells | Standard heterologous host for recombinant protein production [5] [40]. |
| Protein Purification | Ni-NTA Affinity Resin | Immobilized metal affinity chromatography for purifying His-tagged enzymes [5] [40]. |
| Anaerobic Chamber | Glove box with Oâ‚‚ < 2 ppm | Essential for handling and reconstituting oxygen-sensitive metalloenzymes like PolF [5]. |
| Enzyme Cofactors | Fe(II) (e.g., (NH₄)₂Fe(SO₄)₂), SAM, αKG | Required for reconstituting and assaying enzyme activity [5] [40] [41]. |
| Analytical Standards | Polyoximic acid, Azetidine-2-carboxylic acid (AZE) | Authentic compounds for validating product identity via LC-MS and NMR [5] [40]. |
| Derivatization Agent | Dansyl Chloride (DnsCl) | Fluorescent tagging of amino acid products for sensitive LC-MS detection [5]. |
| Crystallography Screen | Commercial Sparse Matrix Screens (e.g., from Hampton Research) | Initial screening for obtaining protein crystals for structural studies [40]. |
Discussion and Biotechnological Outlook
The comparative analysis of these three pathways reveals how nature has converged upon different solutions for azetidine ring formation. The HDO pathway is notable for its radical-based mechanism that directly functionalizes simple amino acids, potentially offering a more straightforward biocatalytic route. The SAM-dependent pathway leverages the intrinsic reactivity of SAM, providing a single-step, atom-economical cyclization. The αKG-dependent pathway exemplifies the versatility of a common oxidative enzyme family in constructing strained C–N bonds.
From a biotechnological perspective, each pathway presents unique advantages and challenges for the production of azetidine-containing compounds or engineering novel analogs. SAM-dependent AZE synthases have been successfully introduced into heterologous biosynthetic pathways to produce analogs like azabicyclenes, demonstrating their potential in combinatorial biosynthesis [40]. However, the metabolic cost of SAM and feedback inhibition by SAH can limit yields [43]. The HDO pathway, with its dependence on readily available amino acid substrates, could offer a more efficient platform if the challenges of handling oxygen-sensitive diiron clusters are managed. Future efforts in enzyme engineering and pathway optimization will be crucial to harnessing these enzymes for the sustainable production of high-value azetidine-based pharmaceuticals and fine chemicals.
The biosynthesis of azetidine-containing amino acids, such as polyoximic acid found in the antifungal agent polyoxin A, represents a significant biochemical challenge due to the high ring strain of the four-membered aza-cycle. Traditional synthetic approaches often require metabolically expensive precursors and harsh conditions. Recent research has unveiled a novel pathway in Streptomyces cacaoi mediated by non-haem iron-dependent enzymes, PolE and PolF, which directly transform proteinogenic amino acids into azetidine derivatives. This whitepaper examines the considerable advantages of this non-haem iron route, focusing on its exceptional catalytic efficiency, minimal precursor requirements, and mechanistic elegance. We present quantitative data, detailed experimental methodologies, and visualizations that underscore the potential of this biosynthetic system for the sustainable production of valuable azetidine-based pharmaceutical intermediates.
Azetidine, a four-membered nitrogen-containing saturated heterocycle, is a crucial structural motif in numerous bioactive compounds and pharmaceutical agents [5]. Its inherent ring strain (approximately 25.4 kcal molâ»Â¹) confers significant synthetic utility but also presents substantial challenges for chemical synthesis [5]. Prior to the elucidation of the non-haem iron route, the known biosynthetic mechanisms for azetidine formation were limited and relied on specialized, high-energy precursors such as S-adenosyl-L-methionine (SAM) or complex radical-mediated rearrangements catalyzed by α-ketoglutarate-dependent oxygenases [5].
The discovery that the enzymes PolE and PolF can directly convert simple, proteinogenic amino acids like L-isoleucine (L-Ile) and L-valine (L-Val) into azetidine amino acids via a non-haem iron-dependent pathway represents a paradigm shift in our understanding of strained heterocycle biosynthesis [5] [27]. This route stands out for its precursor economy, utilizing readily available amino acids, and its catalytic efficiency, employing iron and molecular oxygen as benign cofactors. This guide provides an in-depth technical analysis of these advantages for researchers and drug development professionals working in natural product biosynthesis and biocatalysis.
The Enzymatic Machinery: PolE and PolF
Key Enzyme Functions
The azetidine biosynthesis pathway is primarily driven by two non-haem iron enzymes that operate in a coordinated manner.
- PolF (HDO Superfamily): A haem-oxygenase-like dimetal oxidase (HDO) that constitutes the core catalytic engine. It contains a diiron (Fe₂) center and is sufficient to catalyze the complete transformation of L-Ile and L-Val into their respective azetidine products, polyoximic acid (PA) and 3-methylen-azetidine-2-carboxylic acid (MAA) [5]. Its mechanism involves a potent μ-peroxo-Fe(III)₂ intermediate capable of cleaving unactivated C–H bonds, leading to desaturation, hydroxylation, and radical-mediated C–N bond formation [5].
- PolE (DUF6421 Family): An iron and pterin-dependent oxidase that acts as a metabolic helper enzyme. It specifically catalyzes the 3,4-desaturation of L-Ile, increasing the flux of this intermediate through the PolF-catalyzed cyclization pathway, thereby enhancing the overall titre of the final azetidine product [5] [27].
The table below summarizes the distinct roles and characteristics of these two enzymes.
Table 1: Key Enzymes in the Non-Haem Iron Azetidine Biosynthesis Pathway
| Enzyme | Family | Cofactors | Primary Catalytic Function | Key Intermediate Generated |
|---|---|---|---|---|
| PolF | HDO (Dimetal oxidase/oxygenase) | Diiron (Fe₂), O₂ | Multi-step azetidine ring formation from L-Ile/L-Val | μ-peroxo-Fe(III)₂ |
| PolE | DUF6421 | Fe(II), Pterin | Substrate pre-processing via 3,4-desaturation of L-Ile | 3,4-dehydroisoleucine |
Biosynthetic Pathway Workflow
The following diagram illustrates the coordinated sequence of enzymatic events in the biosynthesis of polyoximic acid from L-isoleucine.
Diagram 1: The Non-Haem Iron Route to Polyoximic Acid. This workflow shows how PolE and PolF act sequentially on L-Ile, with PolF utilizing Oâ‚‚ to form the azetidine ring.
Quantitative Analysis of Efficiency and Specificity
Substrate Scope and Product Profile of PolF
The catalytic prowess of PolF is demonstrated by its ability to handle multiple aliphatic amino acid substrates, forming azetidine rings with high specificity when a β-methyl group is present. The quantitative product distribution under single-turnover conditions provides critical insight into its mechanism and efficiency.
Table 2: Substrate Specificity and Product Formation Catalyzed by PolF
| Substrate | Major Product(s) | Product Type | Key Observation |
|---|---|---|---|
| L-Isoleucine (L-Ile) | Polyoximic Acid (PA) | Azetidine amino acid | Native biosynthetic product; pathway is essential for polyoxin A production [5]. |
| L-Valine (L-Val) | 3-Methylene-azetidine-2-Carboxylic Acid (MAA) | Azetidine amino acid | Demonstrates capability beyond native substrate, highlighting versatility [5]. |
| L-Leucine (L-Leu) | Minor desaturation; Mostly hydroxylation | Hydroxylated amino acid | Absence of azetidine product underscores importance of β-methyl group for cyclization [5]. |
| L-Methionine (L-Met) | Minor desaturation; Mostly hydroxylation | Hydroxylated amino acid | Further evidence that substrate size and structure are critical for C–N bond formation [5]. |
Table 3: Product Distribution in PolF-Catalyzed Transformation of L-Valine (Single Turnover)
| Product | Structural Modification | Formation Rate (minâ»Â¹) | Role in Mechanism |
|---|---|---|---|
| 3,4-dehydrovaline (3,4-dh-Val) | -2 Da (Desaturation) | 0.23 | Major intermediate; precursor to cyclization [5]. |
| 3-dimethylaziridine-2-carboxylic acid (Azi) | -4 Da (Cyclization) | 0.15 | Three-membered N-heterocycle; demonstrates C–N bond forming capability [5]. |
| 4-hydroxyvaline (4-OH-Val) | +16 Da (Hydroxylation) | 0.14 | Competitive shunt product [5]. |
| 3-hydroxyvaline (3-OH-Val) | +16 Da (Hydroxylation) | 0.036 | Minor competitive shunt product [5]. |
Detailed Experimental Protocols
To enable researchers to validate and build upon these findings, this section outlines the key experimental methodologies used to characterize the non-haem iron route.
In Vitro Reconstitution of PolF Activity
Objective: To demonstrate that PolF alone is sufficient to convert L-Ile or L-Val into azetidine products in the presence of Fe(II) and Oâ‚‚.
- Enzyme Preparation: Express and purify PolF from E. coli [5]. The enzyme is initially isolated in an apo (metal-free) form.
- Anaerobic Reconstitution: Incubate apo-PolF with a 3-fold molar excess of Fe(II) under anaerobic conditions (e.g., in an glove box) [5].
- Removal of Excess Iron: Pass the reconstitution mixture through a desalting column (e.g., PD-10) equilibrated with an anaerobic buffer to remove unbound Fe(II). Iron analysis confirms incorporation of approximately 1.5-2.0 equivalents of Fe per PolF monomer [5].
- Reaction Setup: In an anaerobic chamber, mix the reconstituted holo-PolF with substrate (L-Ile or L-Val, 1-5 mM) in a suitable buffer [5].
- Initiation and Quenching: Initiate the reaction by rapidly adding an Oâ‚‚-saturated buffer to the anaerobic mixture. Allow the reaction to proceed for a set time (e.g., 30-60 minutes) before quenching, typically by acidification or flash-freezing [5].
- Product Analysis: Derivatize quenched reaction aliquots with dansyl chloride (DnsCl). Analyze the derivatized products using Liquid Chromatography-Mass Spectrometry (LC-MS). Identify polyoximic acid by its characteristic mass shift of -4 Da relative to L-Ile and confirm by comparison with an authentic standard [5].
Critical Controls: Assays must include controls without enzyme, without Fe(II), and without Oâ‚‚ to confirm the dependence of the reaction on all components [5].
Single Turnover Kinetic Analysis
Objective: To trap and quantify reactive intermediates by limiting the number of available oxidizing equivalents.
- Stoichiometric Fe Binding: Reconstitute apo-PolF with exactly 2 equivalents of Fe(II) per monomer, ensuring no excess reductant is present [5].
- Reaction Initiation: Mix the stoichiometrically reconstituted PolF with substrate and rapidly expose to Oâ‚‚ as described in 4.1.
- Time-Point Sampling: Withdraw aliquots at very short time intervals (e.g., 5, 10, 30, 60 seconds) post-Oâ‚‚ addition and quench immediately [5].
- Quantification: Analyze quenched samples via LC-MS to track the disappearance of the substrate and the appearance and subsequent disappearance of intermediates (e.g., 3,4-dh-Val) and final products (e.g., MAA, Azi, hydroxylated products) [5]. This allows for the calculation of individual formation rates for each species, as shown in Table 3.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogues crucial reagents and materials required for experimental research on non-haem iron-dependent azetidine biosynthesis.
Table 4: Key Research Reagent Solutions for Investigating the Non-Haem Iron Route
| Reagent / Material | Function / Application | Key Notes |
|---|---|---|
| Apo-PolF Enzyme | Core catalyst for in vitro activity assays. | Purified from recombinant E. coli; store in metal-free buffers [5]. |
| Fe(II) Salts (e.g., (NHâ‚„)â‚‚Fe(SOâ‚„)â‚‚) | Cofactor for enzyme reconstitution. | Prepare fresh solutions in degassed, acidic water to prevent oxidation [5]. |
| Oâ‚‚-Saturated Buffer | Oxidant for initiating the catalytic cycle. | Saturate reaction buffer with pure Oâ‚‚ gas prior to use [5]. |
| L-Ile, L-Val, & Analogues | Native and probe substrates. | Used to test substrate specificity and enzyme mechanism [5]. |
| Dansyl Chloride (DnsCl) | Derivatizing agent for LC-MS analysis. | Enhances detection and separation of amino acid substrates and products [5]. |
| Anaerobic Chamber | Provides controlled atmosphere for enzyme reconstitution and reaction setup. | Essential for maintaining the Fe(II) state prior to reaction initiation [5]. |
| Polyoximic Acid Standard | Authentic standard for product verification. | Can be prepared from hydrolyzed polyoxin A for definitive product identification via LC-MS/NMR [5]. |
The non-haem iron route to azetidine amino acids, exemplified by the PolE/PolF system, offers a paradigm of efficiency and precursor economy in natural product biosynthesis. Its primary advantages are threefold:
- Direct Utilization of Simple Precursors: It bypasses the need for expensive, dedicated precursors like SAM, instead leveraging the abundant proteinogenic amino acids L-Ile and L-Val [5].
- Multifunctional Catalytic Power: A single enzyme, PolF, elegantly orchestrates chemically challenging steps—including unactivated C–H bond cleavage and radical-mediated C–N bond formation—using a diiron core and atmospheric oxygen [5].
- Metabolic Efficiency: The helper role of PolE increases pathway flux by generating a desaturated intermediate, optimizing the biosynthetic route for the producer organism and potential biocatalytic applications [5].
For the field of drug development, this elucidated pathway provides a new enzymatic toolkit for the sustainable and stereoselective synthesis of azetidine rings, which are prized in medicinal chemistry for their ability to improve the metabolic stability and target affinity of peptide-based therapeutics. Future research should focus on harnessing these enzymes through protein engineering and synthetic biology to produce novel azetidine-containing compounds with tailored pharmaceutical properties.
Broader Implications for HDO Enzyme Catalysis and Natural Product Biosynthesis
The heme-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily represents an emerging class of metalloenzymes that are redefining our understanding of biological catalysis. These enzymes conserve a flexible protein scaffold shared with heme oxygenase but typically employ dinuclear metal cofactors—most commonly diiron clusters—to activate molecular oxygen for chemically challenging transformations [45]. Unlike the more established ferritin-like diiron oxidases/oxygenases (FDOs), HDOs exhibit distinctive metal-binding properties that allow for dynamic cofactor assembly and disassembly, suggesting a more transient use of iron that may function more as a substrate than a permanent cofactor [45]. This unique feature enables HDOs to catalyze an remarkable array of oxidative reactions, including fragmentation, rearrangement, desaturation, and N-oxygenation processes that remain unprecedented among other dinuclear iron enzymes [45].
The discovery that HDOs mediate the biosynthesis of strained heterocyclic compounds like azetidines represents a paradigm shift in our understanding of natural product biosynthesis. This technical guide examines the broader implications of HDO catalysis through the lens of recent groundbreaking discoveries, with particular emphasis on the enzymatic formation of azetidine amino acids—a transformation that exemplifies the unique catalytic prowess of this enzyme superfamily and its potential to revolutionize synthetic biology and pharmaceutical development.
Structural and Mechanistic Hallmarks of HDO Enzymes
Defining Structural Features
HDO enzymes share a conserved structural scaffold characterized by a seven-helix bundle fold that was first identified in heme oxygenase [45]. Within this architecture, three core helices (α1-α3) house the essential metal-binding residues, while four auxiliary helices provide structural context and substrate-binding determinants [45]. The metal-binding motif typically consists of three histidine and three carboxylate side chains arranged in a distinctive configuration: core helix α1 contributes two ligands (a carboxylate and a histidine), core helix α2 provides a single carboxylate ligand, and core helix α3 supplies three ligands in a characteristic HX₃D/EX₂H motif [45].
This structural organization differs fundamentally from the ferritin-like diiron oxidases/oxygenases (FDOs), which employ a more symmetric ligand arrangement within a four-helix bundle architecture [45]. The HDO scaffold features surface-exposed metal-binding motifs that confer both dynamic metal binding properties and unusual reactivity to its associated metallocofactor [45]. This structural flexibility enables the HDO active site to accommodate diverse substrates and reaction outcomes that are rarely observed in more rigid enzymatic frameworks.
Metal Cofactor Diversity and Assembly
While initially characterized as diiron enzymes, the HDO superfamily exhibits remarkable diversity in metal ion composition and nuclearity. Most characterized HDOs assemble a diiron cluster that reacts with O₂ to form a peroxo-Fe₂(III/III) intermediate, which serves as a common precursor to various reactive species [45]. However, recent studies have revealed exceptions to this paradigm. FlcD, an HDO involved in fluopsin C biosynthesis, utilizes a mononuclear iron cofactor coordinated by two histidines, one glutamate, and substrate-derived ligands [46]. This mononuclear iron center exhibits octahedral geometry reminiscent of the 2-His-1-Glu facial triad in non-heme iron α-ketoglutarate-dependent enzymes but displays meridional geometry [46]. Additionally, CADD (Chlamydia protein associating with death domains) appears to employ a dinuclear Mn/Fe cofactor, further expanding the metallochemical repertoire of HDOs [46].
Table 1: Diversity of Metal Cofactors in Characterized HDO Enzymes
| Enzyme | Metal Cofactor | Reaction Catalyzed | Structural Features |
|---|---|---|---|
| PolF | Diiron (Feâ‚‚) | Azetidine ring formation from L-Ile/L-Val | Conserved E-H-E-H-D-E-H motif [5] |
| FlcD | Mononuclear Fe | Methine excision from oxime substrate | E-H-E-H-V-R motif; octahedral geometry [46] |
| CADD | Mn/Fe heterodinuclear | Tyrosine side-chain cleavage to p-aminobenzoate | Divergent metal coordination sphere [46] |
| UndA | Diiron (Feâ‚‚) | Oxidative decarboxylation to terminal alkene | Substrate-triggered cofactor assembly [47] |
| AetD | Diiron (Feâ‚‚) | Tryptophan to indole nitrile conversion | Substrate-triggered; open coordination site [47] |
Mechanistic Paradigms in HDO Catalysis
HDO enzymes employ diverse mechanistic strategies for O₂ activation and substrate transformation. A key intermediate common to diiron-containing HDOs is the μ-1,2-peroxo-Fe₂(III/III) complex, which forms when the Fe₂(II/II) reactant state reacts with O₂ [45]. This peroxo adopter can then follow several divergent pathways:
Direct substrate oxidation: In PolF, the μ-peroxo-Fe₂(III/III) species directly cleaves unactivated C-H bonds with bond dissociation energies up to 101 kcal/mol, initiating a radical mechanism that culminates in C-N bond formation and azetidine ring closure [5] [10].
Peroxo rearrangement: Some HDOs convert the initial peroxo adduct to geometrically or chemically distinct peroxide-level intermediates capable of reacting with nucleophilic or aromatic substrates [45].
High-valent iron formation: Similar to certain FDOs, some HDOs may generate high-valent Feâ‚‚(III/IV) or Feâ‚‚(IV/IV) intermediates via reductive or homolytic O-O bond cleavage [45].
The dynamic metal binding properties of HDOs also influence their mechanistic behavior. Many HDOs are isolated in apo or partially metallated states and exhibit substrate-triggered cofactor assembly, where substrate binding facilitates metal coordination and active site organization [47]. This contrasts with FDOs, which typically maintain stable, pre-assembled diiron cofactors in their resting states [45].
Figure 1: Divergent Mechanistic Pathways from Common Peroxo Intermediate in HDO Enzymes
Case Study: Azetidine Biosynthesis by PolF and PolE
Discovery and Functional Characterization
The recent elucidation of the azetidine biosynthetic pathway in the polyoxin antifungal producer Streptomyces cacaoi has revealed unprecedented enzymatic capabilities within the HDO superfamily. Gene knockout experiments established that PolF is essential for polyoximic acid (PA) biosynthesis, with polF mutants producing less than 1% of wild-type polyoxin A levels [5] [10]. PolE, while not essential, significantly enhances production efficiency, with polE mutants yielding approximately 10% of wild-type polyoxin A [5] [10].
Functional characterization demonstrated that PolF catalyzes the transformation of L-isoleucine (L-Ile) and L-valine (L-Val) to their corresponding azetidine derivatives via a 3,4-desaturated intermediate [5] [10]. Under multiple turnover conditions with L-Val as substrate, PolF produces 3-methylene-azetidine-2-carboxylic acid (MAA) as the major product, along with several reactive intermediates including 3,4-dehydrovaline (3,4-dhVal) and 3-dimethylaziridine-2-carboxylic acid (Azi) [5]. This remarkable transformation represents the first documented example of an HDO enzyme catalyzing the formation of strained nitrogen-containing heterocycles.
Table 2: Substrate Specificity and Products of PolF-Catalyzed Reactions
| Substrate | Major Product | Additional Products | Proposed Pathway |
|---|---|---|---|
| L-Isoleucine | Polyoximic acid (PA) | Minor hydroxylation products | Azetidine formation via 3,4-desaturation [5] |
| L-Valine | 3-methylene-azetidine-2-carboxylic acid (MAA) | 3,4-dehydrovaline, 3-OH-Val, 4-OH-Val, aziridine intermediate | Azetidine formation with detectable intermediates [5] |
| L-Leucine | Hydroxylation products (major), desaturation products (minor) | N/A | Dominant hydroxylation over cyclization [5] |
| L-Methionine | Hydroxylation products (major), desaturation products (minor) | N/A | Dominant hydroxylation over cyclization [5] |
| L-allo-Ile, D-Ile, D-allo-Ile | Azetidine products (trace amounts) | N/A | Stereochemistry important but not absolute [5] |
Experimental Approaches for HDO Characterization
The study of HDO enzymes requires specialized methodologies to address their dynamic metal binding properties and complex reaction mechanisms. Key experimental approaches include:
1. Anaerobic Protein Reconstitution:
- Purify apo-PolF from E. coli expression systems [5] [10]
- Incubate with excess Fe(II) (typically 3 equivalents) under anaerobic conditions [5] [10]
- Remove unbound Fe(II) using desalting columns [5] [10]
- Typical iron occupancy after reconstitution: ~1.5 equivalents Fe per enzyme [5]
2. Activity Assays Under Controlled Atmosphere:
- Prepare enzyme-substrate mixtures anaerobically [5]
- Initiate reactions by addition of Oâ‚‚-saturated buffer [5]
- Include external reductants (ascorbate or dithiothreitol) for multiple turnover conditions [5] [10]
- For single-turnover studies: use stoichiometric Fe(II) (2 equivalents) without reductants [5]
3. Intermediate Trapping and Analysis:
- Employ single-turnover conditions to accumulate reactive intermediates [5]
- Derivatize products with dansyl chloride (DnsCl) for LC-MS analysis [5]
- Identify intermediates by comparison to authentic standards [5]
- Isolate and characterize products by NMR spectroscopy [5] [10]
4. Spectroscopic Characterization:
- Utilize Mössbauer spectroscopy to identify iron oxidation states and coordination environments [23]
- Apply stopped-flow spectroscopy to monitor rapid reaction kinetics [23]
- Employ electron paramagnetic resonance (EPR) spectroscopy to detect radical intermediates [46]
5. Structural Analysis:
- Determine crystal structures of substrate-bound complexes [5]
- Generate iron anomalous difference maps to identify metal coordination geometry [46]
- Analyze active site architecture to elucidate substrate positioning and catalytic residues [5] [47]
Figure 2: Experimental Workflow for Characterization of Novel HDO Enzymes
The Scientist's Toolkit: Essential Reagents and Methods
Table 3: Key Research Reagent Solutions for HDO Enzyme Studies
| Reagent/Method | Function/Application | Technical Considerations |
|---|---|---|
| Anaerobic Chamber | Maintain oxygen-free environment for metal reconstitution | Critical for preserving Fe(II) oxidation state [5] |
| Fe(II) Salts (e.g., FeSOâ‚„) | Cofactor reconstitution | Use fresh solutions; 3 eq. typically required for full occupancy [5] |
| Ascorbate/DTT | External reductant for multiple turnover | Regenerates Fe(II) state during catalytic cycling [5] [10] |
| Oâ‚‚-Saturated Buffer | Initiate oxidative reactions | Prepared by bubbling Oâ‚‚ through buffer; add to start reactions [5] |
| Dansyl Chloride (DnsCl) | Derivatization for LC-MS detection | Enhances detection sensitivity for amino acid products [5] |
| Mössbauer Spectroscopy | Probe iron oxidation states and coordination | Requires âµâ·Fe-enriched samples for highest resolution [23] [46] |
| Stopped-Flow Spectroscopy | Monitor rapid reaction kinetics | Millisecond time resolution for intermediate detection [23] |
| X-ray Crystallography | Structural determination of metal centers | Often requires anaerobic crystallization; substrate soaking [5] [46] |
Implications for Natural Product Biosynthesis and Drug Development
Expanding the Reaction Landscape of Non-Heme Iron Enzymes
The discovery of azetidine-forming activity in PolF substantially expands the known catalytic repertoire of non-heme iron enzymes and the HDO superfamily specifically. Prior to this finding, HDOs were known to catalyze N-oxygenation (SznF, RohS), desaturase-lyase reactions (UndA, BesC), methine excision (FlcD), and oxidative rearrangements (AetD) [46] [47]. The ability of PolF to construct strained four-membered azetidine rings—and even three-membered aziridine rings—introduces a new dimension of complexity to HDO catalysis [23]. This finding suggests that the structural flexibility of the HDO active site enables the stabilization of high-energy transition states involved in small-ring formation, a capability with profound implications for natural product biosynthesis.
The HDO superfamily represents a rapidly expanding group of enzymes with largely unexplored sequence space. Bioinformatic analyses indicate that thousands of uncharacterized HDO homologs exist in microbial genomes, suggesting that additional novel reaction types remain to be discovered [45] [46]. The dynamic metal binding properties and structural plasticity of these enzymes may underlie their functional diversity, positioning the HDO superfamily as a rich resource for discovering new biocatalysts.
Biocatalytic Applications in Pharmaceutical Synthesis
The enzymatic synthesis of azetidine rings by PolF and related HDO enzymes has significant implications for pharmaceutical manufacturing. Azetidine-containing compounds exhibit diverse bioactivities, including antibiotic, antiviral, and anticancer properties [5] [7]. Traditional chemical synthesis of these strained heterocycles requires harsh conditions, expensive catalysts, and often suffers from poor stereocontrol [23]. The PolF-catalyzed route employs an inexpensive amino acid precursor (L-isoleucine) and functions in aqueous solution under mild conditions, offering a sustainable alternative to conventional synthetic approaches [23].
The substrate promiscuity of PolF further enhances its biocatalytic potential. The enzyme accepts both L-Ile and L-Val as substrates, producing structurally diverse azetidine products [5]. This flexibility suggests opportunities for engineering PolF variants with expanded substrate scope, potentially enabling the enzymatic synthesis of non-proteinogenic azetidine amino acids for peptide-based therapeutics. The discovery that PolF can also produce aziridine (three-membered) rings further increases its synthetic utility [23].
Functional and Evolutionary Insights
The existence of two distinct enzymes (PolF and PolE) involved in azetidine biosynthesis raises intriguing questions about the evolutionary drivers shaping metabolic pathways. PolE, an Fe- and pterin-dependent oxidase, catalyzes the desaturation of L-Ile, enhancing the flux through the azetidine biosynthetic pathway [5] [10]. This functional redundancy or enhancement may reflect evolutionary optimization for metabolic efficiency or regulatory control in natural producers.
The HDO superfamily exhibits fascinating evolutionary relationships, with members like FlcD employing mononuclear iron centers despite sharing the canonical heme oxygenase fold [46]. This structural conservation with functional divergence suggests that the HDO scaffold provides an optimal framework for evolving new catalytic activities through modifications of metal coordination spheres and active site contours. The dynamic nature of the metal-binding motifs in HDOs may facilitate evolutionary exploration of new chemical space by enabling rapid adaptation of metallocofactor properties.
Future Directions and Research Opportunities
The emerging understanding of HDO enzymology opens numerous avenues for future research. Key areas include:
Exploration of HDO sequence space: Systematic characterization of unstudied HDO homologs will likely reveal new reaction types and expand our understanding of the catalytic potential of this superfamily.
Protein engineering for biocatalytic applications: Rational design and directed evolution of HDO enzymes like PolF could yield optimized catalysts for pharmaceutical synthesis with enhanced activity, stability, and substrate scope.
Mechanistic studies of underrepresented HDO subtypes: Further investigation of mononuclear and heterodinuclear HDOs will provide comparative insights into structure-function relationships across the superfamily.
Integration of HDO catalysis into metabolic engineering platforms: Developing microbial hosts for heterologous expression of HDO pathways could enable sustainable production of valuable azetidine-containing compounds.
The discovery of azetidine biosynthesis by HDO enzymes represents a watershed moment in natural product research, highlighting nature's sophisticated solutions to chemically challenging transformations. As research in this field advances, the HDO superfamily will undoubtedly yield additional surprises and opportunities for innovation in synthetic biology, drug discovery, and green chemistry.
Azetidine, a saturated four-membered nitrogen-containing heterocycle, has emerged as a crucial structural motif in medicinal chemistry due to its significant ring strain (approximately 25.4 kcal molâ»Â¹) and unique physicochemical properties. [5] This strained ring system is present in dozens of drug-related molecules of both natural and synthetic origins, conferring enhanced target binding affinity, metabolic stability, and desirable pharmacokinetic profiles. [48] The azetidine ring is found in many compounds with important bioactivity, including the naturally occurring antifungal agent polyoxin A, which contains an azetidine amino acid called polyoximic acid (PA). [5] For decades, the biosynthetic pathways responsible for azetidine formation in natural products remained enigmatic, limiting our ability to harness these pathways for drug development. Recent groundbreaking research has finally elucidated the novel enzymatic machinery behind azetidine biosynthesis, revealing an unexpected dependence on non-haem iron-dependent enzymes. [5] [48] This discovery opens unprecedented opportunities for antifungal drug development and beyond, promising to revolutionize our approach to designing next-generation therapeutic agents.
The pressing need for novel antifungal compounds has gained urgent attention due to increasing incidences of fungal infections among immunocompromised patients, including those with COVID-19. [49] Worldwide, more than 1 billion people are infected with fungal infections, resulting in 1.5-2 million deaths annually. [49] The development of antifungal agents based on azetidine-containing compounds represents a promising strategy to address this growing public health threat, particularly in an era of increasing antimicrobial resistance. This technical review comprehensively examines the therapeutic implications of the newly discovered azetidine biosynthesis pathways, with a specific focus on their potential for antifungal drug development and other biomedical applications.
Novel Biosynthetic Pathways for Azetidine Formation
The PolE-PolF Enzymatic Cascade in Polyoximic Acid Biosynthesis
Recent research has elucidated a novel two-metalloenzyme cascade responsible for constructing the azetidine-containing pharmacophore in natural products. [48] This pathway centers on two key enzymes, PolE and PolF, which work in concert to transform proteinogenic amino acids into azetidine-containing compounds:
PolE Function and Mechanism: PolE, a member of the DUF6421 family, has been identified as an Fe²⺠and pterin-dependent oxidase that catalyzes the desaturation of L-isoleucine (L-Ile). [5] [48] This enzyme introduces a double bond between the C3 and C4 atoms of L-Ile, creating a 3,4-desaturated intermediate that serves as a substrate for the subsequent cyclization reaction.
PolF Function and Mechanism: PolF is a member of the haem-oxygenase-like dimetal oxidase and/or oxygenase (HDO) superfamily. [5] Remarkably, this enzyme alone is sufficient for the transformation of L-Ile and L-valine to their azetidine derivatives via a 3,4-desaturated intermediate. [5] PolF exhibits dual functionality, orchestrating both the desaturation and cyclization reactions with L-Ile as the initial substrate. [48]
The collaborative action of these enzymes significantly increases the flux of L-Ile desaturation, making the biosynthetic pathway both specific and efficient. [5] Genetic experiments have confirmed the essential nature of these enzymes, with polF knockout mutants failing to produce any measurable polyoxin A (<1% of wild-type), while polE mutants showed reduced but detectable amounts (~10% of wild-type). [5]
Mechanistic Insights into Azetidine Ring Formation
The mechanism of azetidine ring formation by PolF represents a remarkable example of enzymatic catalysis. Mechanistic studies indicate that a μ-peroxo-Fe(III)₂ intermediate is directly responsible for unactivated C–H bond cleavage, with post-H-abstraction reactions (including the critical C–N bond formation) proceeding through radical mechanisms. [5] Structural analyses using X-ray crystallography have provided vivid snapshots of the enzyme active sites, illustrating the delicate interplay between substrate positioning and iron coordination environment required to promote azetidine ring closure. [33]
Table 1: Key Enzymes in Azetidine Amino Acid Biosynthesis
| Enzyme | Family | Cofactors | Catalytic Function | Products |
|---|---|---|---|---|
| PolE | DUF6421 | Fe²âº, pterin | L-isoleucine desaturase | 3,4-desaturated L-Ile intermediate |
| PolF | HDO superfamily | Diiron center | Dual-function oxidase/cyclase | Polyoximic acid from L-Ile; 3-methyl-ene-azetidine-2-carboxylic acid from L-Val |
The PolF enzyme demonstrates interesting substrate specificity, showing highest activity toward medium-size aliphatic amino acids. While L-Ile and L-valine yield azetidine products, other amino acids like L-leucine and L-methionine primarily yield hydroxylation products with only minor desaturation products. [5] This specificity suggests that the β-methyl group is critical for azetidine formation, providing important insights for designing enzyme inhibitors or engineering substrate specificity for biotechnological applications.
Experimental Approaches for Studying Azetidine Biosynthesis
Key Methodologies and Workflows
Elucidating the azetidine biosynthetic pathway required the application of cutting-edge genomic, biochemical, and structural biology techniques. The following workflow diagram illustrates the integrated experimental approach used to characterize the PolE-PolF enzymatic cascade:
Essential Research Reagents and Solutions
The following table comprehensively details the key reagents, materials, and instrumentation required for investigating azetidine biosynthesis pathways, based on methodologies described in the recent literature:
Table 2: Essential Research Reagents for Azetidine Biosynthesis Studies
| Reagent/Material | Specification/Example | Experimental Function | Reference Application |
|---|---|---|---|
| PolE & PolF Enzymes | Recombinant proteins from E. coli expression | Catalytic components for in vitro assays | [5] |
| Amino Acid Substrates | L-isoleucine, L-valine, analogues | Enzyme substrates for activity assays | [5] |
| Cofactors | Fe²⺠(FeSO₄/FeCl₂), pterin | Essential metalloenzyme cofactors | [5] [48] |
| Analytical Standards | Polyoximic acid, 3,4-dehydrovaline | LC-MS calibration and product identification | [5] |
| Derivatization Reagent | Dansyl chloride (DnsCl) | Product derivatization for detection | [5] |
| Chromatography System | LC-MS with C18 column | Separation and identification of reaction products | [5] |
| Structural Biology Tools | X-ray crystallography, cryo-EM | Enzyme-substrate complex structure determination | [33] [48] |
Detailed Enzymatic Assay Protocols
For researchers seeking to replicate or build upon these findings, the following detailed protocols for in vitro enzymatic characterization have been extracted from the primary literature:
PolF Activity Assay: Apo-PolF is initially reconstituted with excess Fe(II) (3 equivalents) under anaerobic conditions in an inert atmosphere glovebox. After removal of unbound Fe(II) using a desalting column, the enzyme (typically 5-10 μM) is incubated with substrate (L-Ile or L-Val, 100-500 μM) in anaerobic buffer. Reactions are initiated by adding O₂-saturated buffer and proceeded with gentle agitation at room temperature. For multiple turnover conditions, an external reductant such as ascorbate (1-5 mM) or dithiothreitol is included to regenerate the diiron center. [5]
Product Analysis: Reaction products are derivatized with dansyl chloride (DnsCl) for enhanced detection sensitivity. Analysis is performed via LC-MS with comparison to authentic standards. For polyoximic acid identification, the authentic standard can be prepared from purified polyoxin A through acid hydrolysis. [5] Under single turnover conditions (2 equivalents Fe(II) without reductant), reaction intermediates can be trapped and characterized, including 3,4-dehydrovaline (3,4-dh-Val) and 3-dimethylaziridine-2-carboxylic acid (Azi). [5]
Kinetic Analysis: Initial rates are determined under multiple turnover conditions with varying substrate concentrations. For PolF with L-Val as substrate, the major product 3-methylene-azetidine-2-carboxylic acid (MAA) formation follows Michaelis-Menten kinetics, with additional minor products (3-OH-Val, 4-OH-Val, 3,4-dh-Val, Azi) providing mechanistic insights into the parallel pathways. [5]
Implications for Antifungal Drug Development
Azetidine-Containing Natural Products as Antifungal Agents
The azetidine-containing polyoxin family represents well-established antifungal agents that inhibit chitin synthase in pathogenic fungi. [5] The discovery of the biosynthetic pathway for their azetidine moiety opens new avenues for engineering enhanced analogues through metabolic engineering or combinatorial biosynthesis. Additionally, recent research has demonstrated that synthetic azetidine derivatives exhibit significant antifungal activity. A 2023 study reported the synthesis of a novel chitosan-azetidine derivative that showed enhanced antifungal activity against Aspergillus fumigatus 3007 with an antifungal inhibitory index of 26.19%. [49] Confocal and SEM microscopy revealed significant morphological alterations in fungal mycelia treated with this compound, demonstrating its potent antifungal effects. [49]
Strategic Advantages for Antifungal Development
Targeting amino acid biosynthesis pathways represents a validated strategy for antifungal development, as fungi can synthesize all proteinogenic amino acids, including the nine essential for humans. [50] Several enzymes in these biosynthetic pathways are fungi-specific and absent from mammalian cells, making them attractive targets for selective antimicrobial chemotherapy. [50] The discovery of the PolE-PolF pathway offers several strategic advantages:
Novel Mechanism of Action: Azetidine-containing compounds interfere with cellular processes through mechanisms distinct from conventional antifungals, potentially overcoming existing resistance.
Bioinspiration for Drug Design: The enzymatic machinery for azetidine formation provides templates for designing novel inhibitors that target similar pathways in pathogenic fungi.
Biosynthetic Engineering Potential: The genes encoding PolE and PolF can be harnessed for engineered biosynthesis of novel azetidine-containing compounds with enhanced pharmacological properties.
Table 3: Azetidine-Containing Compounds with Antimicrobial Potential
| Compound | Source | Biosynthetic Pathway | Antimicrobial Activity |
|---|---|---|---|
| Polyoxin A | Streptomyces cacaoi | PolE-PolF cascade | Fungicide (chitin synthase inhibitor) |
| Azetidine-2-carboxylic acid | Various plants | Unknown (non-ribosomal) | Antibacterial, antifungal via misincorporation |
| Chitosan-azetidine derivative | Synthetic | Chemical synthesis | Antifungal vs. Aspergillus fumigatus (26.19% inhibition) |
Overcoming Antimicrobial Resistance
The escalating crisis of antimicrobial resistance demands innovative approaches to drug development. Plant-derived amino acid analogues have shown promise as antimicrobial agents because they exhibit unique mechanisms of action that are less susceptible to conventional resistance development. [51] Azetidine-2-carboxylic acid, a plant analogue of proline, exemplifies this strategy by being misincorporated into proteins, causing protein misfiling and dysfunction. [51] This mechanism disrupts multiple cellular processes simultaneously, making it difficult for pathogens to develop resistance through single mutations.
Combination therapies represent another promising application. Research suggests that the antimicrobial activity of amino acid analogues can be enhanced by concomitant starvation for the corresponding standard amino acid. [51] This approach could potentially restore sensitivity to conventional antifungals when used in combination therapy, providing a strategy to overcome multidrug resistance.
Future Directions and Research Opportunities
Enzyme Engineering and Synthetic Biology Applications
The structural and mechanistic insights into PolE and PolF provide a foundation for enzyme engineering efforts to expand the substrate scope and catalytic efficiency of these azetidine-forming enzymes. [33] Protein engineering approaches, including directed evolution and rational design, could generate enzyme variants capable of producing novel azetidine derivatives with enhanced pharmaceutical properties. The crystal structures of PolF in complex with substrates provide essential guidance for these engineering efforts by revealing the molecular determinants of substrate specificity and catalysis. [5]
Synthetic biology approaches offer exciting opportunities to harness the PolE-PolF pathway for heterologous production of azetidine-containing compounds. Reconstruction of the pathway in industrially relevant microbial hosts could enable sustainable production of polyoxin analogues and novel azetidine scaffolds. The modular nature of the two-enzyme system facilitates its integration with other biosynthetic pathways to create hybrid molecules with potentially improved bioactivity.
Expansion to Other Therapeutic Areas
While antifungal applications represent the most immediate therapeutic opportunity, azetidine-containing compounds show promise across multiple therapeutic areas:
Anticancer Applications: The selective toxicity of amino acid analogues to rapidly dividing cells provides a rationale for developing azetidine-containing compounds as anticancer agents. [51] Cancer cells with elevated amino acid transport may be particularly vulnerable to such analogues.
Antibacterial Agents: The unique mechanism of azetidine-containing compounds could address the critical need for novel antibacterial agents, especially against multidrug-resistant Gram-negative pathogens.
Neurological Disorders: The ability of azetidine analogues to induce controlled proteotoxic stress and endoplasmic reticulum stress [51] offers research tools for investigating protein misfolding diseases, potentially leading to novel therapeutic strategies.
The following diagram illustrates the integrated research and development pipeline for translating azetidine biosynthesis discoveries into therapeutic applications:
The recent elucidation of azetidine amino acid biosynthesis by non-haem iron-dependent enzymes represents a transformative advance in natural product biosynthesis and medicinal chemistry. The discovery that PolE and PolF form a two-metalloenzyme cascade capable of constructing the strained azetidine ring system provides both fundamental scientific insights and practical applications for drug development. These findings substantially expand the catalytic repertoire known to non-haem iron enzymes and challenge previous assumptions about the energetic barriers to four-membered ring formation in biological systems.
From a therapeutic perspective, these discoveries arrive at a critical juncture in antimicrobial development, as resistance to existing agents continues to escalate while the pipeline of new compounds has stagnated. The azetidine pharmacophore offers unique opportunities for addressing this challenge through multiple mechanisms, including direct antimicrobial activity, enhancement of existing agents in combination therapy, and novel target engagement strategies. The experimental protocols, reagent resources, and strategic frameworks presented in this review provide researchers with the essential tools to build upon these discoveries and accelerate the development of novel therapeutics based on azetidine chemistry.
As the field progresses, integration of structural biology, enzyme engineering, and synthetic biology will be essential for fully realizing the potential of these findings. The azetidine biosynthetic pathway represents not just a scientific curiosity but a versatile platform for generating molecular diversity with significant implications for antifungal drug development and beyond. By leveraging nature's enzymatic ingenuity, researchers can now access complex ring systems that have challenged synthetic chemists for decades, opening new frontiers in medicinal chemistry and therapeutic development.
Conclusion
The elucidation of the non-haem iron-dependent pathway for azetidine amino acid biosynthesis represents a paradigm shift in our understanding of how nature constructs highly strained heterocycles. The catalytic prowess of PolF, facilitated by PolE, demonstrates an efficient, radical-based mechanism that overcomes the energetic challenges of ring formation without relying on metabolically expensive precursors. This breakthrough not only solves a long-standing biosynthetic enigma but also significantly expands the known functional repertoire of the HDO enzyme superfamily. For biomedical and clinical research, this discovery opens unprecedented avenues. It provides a sustainable, biocatalytic platform for synthesizing azetidine-containing compounds, which are prized in medicinal chemistry for their ability to modulate the properties of therapeutic peptides and small molecules. Future directions should focus on exploiting this pathway for the engineered biosynthesis of novel azetidine-based antifungals and other pharmaceuticals, harnessing directed evolution to broaden substrate scope, and further exploring the role of these unique amino acids in microbial physiology and pathogenesis.
References
- 1. Structurally divergent reactivity of 2,2-disubstituted azetidines [https://pubs.rsc.org/en/content/articlehtml/2025/qo/d5qo00804b]
- 2. azetidine, thiazetidine and oxazetidine [https://www.sciencedirect.com/science/article/abs/pii/S004040392500382X]
- 3. A modular synthesis of azetidines from reactive triplet imine ... [https://www.nature.com/articles/s41929-025-01405-7]
- 4. Recent advances in the synthesis of azetidines [https://www.sciencedirect.com/science/article/abs/pii/S0040402024005647]
- 5. Azetidine amino acid biosynthesis by non-haem iron- ... [https://www.nature.com/articles/s41557-025-01958-x]
- 6. Azetidine amino acid biosynthesis by non-haem iron ... [https://pubmed.ncbi.nlm.nih.gov/41120677/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=08zEbX_nNuHk8qjoqZgyfJkiZZh0ILoW65URJHgQckS&fc=None&ff=20251028055624&v=2.18.0.post22+67771e2]
- 7. Non-Haem Iron Enzymes Drive Azetidine Biosynthesis [https://bioengineer.org/non-haem-iron-enzymes-drive-azetidine-biosynthesis/]
- 8. Modular Access to N-SF5 azetidines - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12443333/]
- 9. Enantioselective Synthesis of 2,3-Disubstituted Azetidines ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12257529/]
- 10. Azetidine amino acid biosynthesis by non-heme iron- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12614342/]
- 11. A route to azetidine | Nature Chemical Biology [https://www.nature.com/articles/s41589-025-02090-0]
- 12. sciencedirect.com/topics/neuroscience/ polyoxin [https://www.sciencedirect.com/topics/neuroscience/polyoxin]
- 13. BGC0000877 [https://mibig.secondarymetabolites.org/repository/BGC0000877.5/index.html]
- 14. Advancing time-resolved structural biology [https://www.nature.com/articles/s41592-025-02659-6]
- 15. Automated Multiconformer Model Building for X- ray ... [https://elifesciences.org/reviewed-preprints/90606]
- 16. X-rays in the Cryo-EM Era: Structural Biology's Dynamic Future [https://pmc.ncbi.nlm.nih.gov/articles/PMC5999524/]
- 17. Gene knockout [https://en.wikipedia.org/wiki/Gene_knockout]
- 18. Gene Knockout | IDT [https://www.idtdna.com/pages/applications/gene-knockout]
- 19. Strategies of genome editing design for gene knockout - NCBI [https://www.ncbi.nlm.nih.gov/books/n/glycopodv2/g122-genomeediting/]
- 20. Enzyme Characterization Methods [https://www.mtoz-biolabs.com/enzyme-characterization-methods.html]
- 21. Functional Characterization of Enzymatic Steps Involved in ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2018.01356/full]
- 22. 9PRR: X-ray crystal structure of Streptomyces cacaoi PolF ... [https://www.rcsb.org/structure/9PRR]
- 23. New enzymes promise cheaper, cleaner drug production [https://medschool.duke.edu/news/new-enzymes-promise-cheaper-cleaner-drug-production]
- 24. A growing family of O2 activating dinuclear iron enzymes ... [https://www.sciencedirect.com/science/article/abs/pii/S0010854516300546]
- 25. Substrate-triggered Formation of a Peroxo-Fe2(III/III) ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6800569/]
- 26. Generation of a μ-1,2-hydroperoxo Fe III ... [https://www.nature.com/articles/s41467-022-28894-5]
- 27. Azetidine amino acid biosynthesis by non-haem iron- ... [https://read.qxmd.com/read/41120677/azetidine-amino-acid-biosynthesis-by-non-haem-iron-dependent-enzymes]
- 28. Functionalized azetidines via visible light-enabled aza ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6841681/]
- 29. Molecular basis for azetidine-2-carboxylic acid biosynthesis [https://pmc.ncbi.nlm.nih.gov/articles/PMC11794875/]
- 30. A sustainable and chemoselective continuous flow ... [https://www.sciencedirect.com/science/article/pii/S277322312300002X]
- 31. Two-Metal Enzyme Cascade Builds Azetidine ... [https://bioengineer.org/two-metal-enzyme-cascade-builds-azetidine-pharmacophore/]
- 32. by Azetidine - amino -dependent... acid biosynthesis non haem iron [https://pubmed.ncbi.nlm.nih.gov/41120677/]
- 33. - Non Drive Haem Iron Enzymes Azetidine Biosynthesis [https://scienmag.com/non-haem-iron-enzymes-drive-azetidine-biosynthesis/]
- 34. Computational stabilization of a non-heme iron enzyme ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11290999/]
- 35. Molecular basis for azetidine-2-carboxylic acid biosynthesis [https://www.nature.com/articles/s41467-025-56610-6]
- 36. Structure-function correlations in oxygen activating non- ... [https://pubmed.ncbi.nlm.nih.gov/16317455/]
- 37. 125 Te and 57 Fe nuclear resonance vibrational ... [https://www.nature.com/articles/s41467-025-62118-w]
- 38. Identification and Spectroscopic Characterization of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4955228/]
- 39. Electronic structures and spectroscopic signatures of diiron ... [https://pubs.rsc.org/en/content/articlelanding/2021/dt/d1dt02286e]
- 40. Molecular basis for azetidine -2-carboxylic acid biosynthesis [https://www.nature.com/articles/s41467-025-56610-6?error=cookies_not_supported&code=0d8c4f05-5d2d-4aa6-b5fe-fcee56566b52]
- 41. Biosynthesis of unique natural product scaffolds by Fe(II) ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11880133/]
- 42. Molecular basis for azetidine -2-carboxylic acid biosynthesis [https://pubmed.ncbi.nlm.nih.gov/39905070/]
- 43. Biotechnological applications of S-adenosyl-methionine- dependent ... [https://bioresourcesbioprocessing.springeropen.com/articles/10.1186/s40643-021-00425-y]
- 44. Recent Examples of α-Ketoglutarate-Dependent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6093783/]
- 45. Heme-oxygenase-like metalloenzymes - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12335807/]
- 46. Structural Basis for Methine Excision by a Heme Oxygenase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11363339/]
- 47. A single diiron enzyme catalyses the oxidative ... [https://www.nature.com/articles/s41557-024-01603-z]
- 48. A two-metalloenzyme cascade constructs the azetidine -containing... [https://www.nature.com/articles/s41557-025-01949-y?error=cookies_not_supported&code=4d9ea770-d868-43c2-bf11-4b0d259fdbc7]
- 49. Antifungal activity of novel azetidine tethered chitosan ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10266883/]
- 50. Inhibitors of amino acids biosynthesis as antifungal agents [https://pmc.ncbi.nlm.nih.gov/articles/PMC4302243/]
- 51. Plant amino acid analogues as antimicrobial agents - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12546413/]