Measuring Success: Key Efficiency Metrics and Optimization Strategies for Biosynthetic Pathways
This comprehensive review addresses the critical challenge of quantifying and enhancing efficiency in biosynthetic pathways for researchers, scientists, and drug development professionals.
Measuring Success: Key Efficiency Metrics and Optimization Strategies for Biosynthetic Pathways
Abstract
This comprehensive review addresses the critical challenge of quantifying and enhancing efficiency in biosynthetic pathways for researchers, scientists, and drug development professionals. We explore foundational metrics like titer, yield, and productivity, then delve into advanced computational methodologies for pathway design and optimization. The article provides practical troubleshooting frameworks for overcoming metabolic bottlenecks and presents rigorous validation approaches through comparative omics analysis. By synthesizing recent advances in lifespan engineering, computational workflow integration, and AI-driven pathway navigation, this resource offers a strategic roadmap for developing high-performance microbial cell factories capable of economically viable production of valuable plant natural products and pharmaceuticals.
Defining Biosynthetic Efficiency: Core Metrics and Fundamental Barriers
In the field of synthetic biology and metabolic engineering, the successful scaling of microbial production from laboratory experiments to industrial manufacturing depends on the rigorous optimization of three fundamental efficiency indicators: titer, yield, and productivity. Collectively known as the TRY metrics, these parameters provide a comprehensive framework for evaluating the technical and economic viability of biosynthetic pathways [1] [2]. Titer, measured typically in grams per liter (g/L), represents the final concentration of the target compound achieved in a fermentation broth, directly influencing downstream processing costs. Yield, expressed as grams of product per gram of substrate (g/g), quantifies the conversion efficiency of raw materials, determining resource utilization and material costs. Productivity, measured as grams per liter per hour (g/L/h), reflects the volumetric production rate, which dictates the reactor size and capital investment required for a given output [1] [3] [2].
The critical importance of these metrics extends beyond technical performance to encompass fundamental economic considerations. As noted in research on strain design strategies, "the economic viability of a bioprocess is commonly evaluated by its product yield, titer, and productivity" [3]. These parameters respectively reflect the downstream processing costs, reactor size determinants, and raw material utilization efficiency that collectively determine commercial feasibility [1]. This guide provides a comparative analysis of TRY metrics across diverse biosynthetic pathways, experimental methodologies for their optimization, and visual frameworks for understanding their interconnected relationships in pathway engineering.
Comparative Performance of TRY Metrics Across Biosynthetic Pathways
The TRY metrics vary significantly across different microbial hosts and target compounds, reflecting the unique metabolic challenges and engineering solutions for each system. The following table summarizes reported performance data for several biologically-produced compounds, illustrating the range of achievable efficiencies.
Table 1: Comparative TRY Metrics for Selected Biological Productions
| Compound | Host Organism | Titer (g/L) | Yield (g/g) | Productivity (g/L/h) | Reference |
|---|---|---|---|---|---|
| Dopamine | E. coli W3110 | 22.58 | - | - | [4] |
| Psilocybin | E. coli (de novo) | 2.00 | - | - | [5] |
| Psilocybin | S. cerevisiae | 0.627 | - | - | [5] |
| Naringenin | E. coli M-PAR-121 | 0.765 | - | - | [6] |
| Naringenin | S. cerevisiae | 1.129 | - | - | [6] |
| Indigoidine | P. putida KT2440 | 25.6 | 0.33 (g/g glucose) | 0.22 | [2] |
The data reveals substantial variability in optimization performance across different host systems. For instance, the highest reported naringenin titer in S. cerevisiae (1.129 g/L) significantly exceeds that in E. coli (0.765 g/L), highlighting host-specific metabolic capabilities [6]. Similarly, psilocybin production has been more successful in S. cerevisiae (627 mg/L in fed-batch) compared to early E. coli systems (27.7 mg/L), though recent engineering advances in E. coli have dramatically improved performance to 2.00 g/L [5]. These differences underscore the importance of host selection and pathway optimization in achieving competitive TRY metrics.
The MCF2Chem knowledge base, a manually curated resource containing 8,888 production records for 1,231 compounds produced by 590 microbial cell factories, provides broader context for these performance benchmarks [7]. Statistical analysis of this database shows that bacteria account for approximately 60% of microbial chassis used in production, with Escherichia coli, Saccharomyces cerevisiae, Yarrowia lipolytica, and Corynebacterium glutamicum collectively synthesizing 78% of reported chemical compounds [7]. This distribution reflects the established engineering tools and metabolic capabilities of these preferred platforms.
Experimental Protocols for TRY Metric Optimization
Systematic Pathway Optimization for Naringenin Production
The stepwise optimization of naringenin production in E. coli demonstrates a systematic methodology for enhancing TRY metrics [6]. The research began with the evaluation of tyrosine ammonia-lyase (TAL) genes from different sources expressed in three distinct E. coli strains to maximize p-coumaric acid production (achieving 2.54 g/L in the tyrosine-overproducing M-PAR-121 strain with TAL from Flavobacterium johnsoniae). The optimal strain was then used to express combinations of 4-coumarate-CoA ligase (4CL) and chalcone synthase (CHS) genes from various organisms, resulting in 560.2 mg/L of naringenin chalcone with the FjTAL, At4CL (Arabidopsis thaliana), and CmCHS (Cucurbita maxima) combination. Finally, different chalcone isomerase (CHI) genes were validated, with CHI from Medicago sativa yielding the highest naringenin production of 765.9 mg/L [6]. This sequential approach isolates variables at each pathway step, enabling identification of the optimal enzyme combination.
Dynamic Strain Scanning Optimization (DySScO) Strategy
For more sophisticated TRY optimization, the Dynamic Strain Scanning Optimization (DySScO) strategy integrates dynamic Flux Balance Analysis (dFBA) with existing strain design algorithms to balance yield, titer, and productivity [3]. This computational framework consists of three phases:
- Scanning Phase: Identification of the production envelope (Pareto frontier in product flux vs. biomass flux) and creation of hypothetical flux distributions along this envelope, followed by dFBA simulations of these distributions in bioreactor environments.
- Design Phase: Application of strain design algorithms (such as OptKnock or GDLS) to find high-product-yield strains within the optimal growth rate range identified in the scanning phase.
- Selection Phase: Dynamic simulation of designed strains using dFBA, performance evaluation using a consolidated performance metric (CSP) that weights yield, titer, and productivity, and selection of the optimal strain design [3].
This approach addresses a critical limitation of metabolic engineering strategies that focus solely on cellular metabolism without considering bioprocess dynamics, thereby enabling simultaneous optimization of all three TRY metrics [3].
Growth-Coupled Production Using Minimal Cut Sets
The application of Minimal Cut Set (MCS) analysis represents an advanced strategy for TRY optimization by genetically rewiring metabolism to couple product synthesis with growth [2]. In one demonstration, researchers computed MCS solution-sets for indigoidine production in Pseudomonas putida KT2440, identifying one experimentally feasible solution requiring 14 simultaneous reaction interventions from 63 possible solutions. Implementing these 14 gene knockdowns using multiplex-CRISPRi shifted production from stationary to exponential phase, achieving 25.6 g/L titer, 0.22 g/L/h productivity, and approximately 50% of the maximum theoretical yield (0.33 g indigoidine/g glucose) [2]. This growth-coupled approach ensures continuous production during active biomass accumulation, significantly enhancing volumetric productivity.
Table 2: Key Research Reagent Solutions for TRY Optimization
| Reagent/Technique | Function in TRY Optimization | Application Example |
|---|---|---|
| Multiplex CRISPRi | Enables simultaneous knockdown of multiple metabolic reactions | Implementing 14 reaction interventions in P. putida for growth-coupled indigoidine production [2] |
| dFBA (Dynamic Flux Balance Analysis) | Models metabolic network within bioreactor dynamics | Predicting titer and productivity in DySScO strategy [3] |
| Minimal Cut Set (MCS) Algorithm | Identifies minimal reaction sets whose elimination couples production to growth | Designing P. putida strain with obligatory indigoidine production during growth [2] |
| Tyrosine-overproducing Strains (E. coli M-PAR-121) | Provides enhanced precursor supply for pathway optimization | Increasing p-coumaric acid production for naringenin synthesis [6] |
| Two-stage pH Fermentation Strategy | Separates growth and production phases, reduces product degradation | Enhancing dopamine yield in E. coli (22.58 g/L) [4] |
Visualization of TRY Optimization Workflows and Metabolic Relationships
The following diagrams illustrate key experimental workflows and metabolic relationships for TRY optimization, providing visual guidance for implementing these strategies.
Diagram 1: A generalized workflow for systematic TRY metric optimization in biosynthetic pathway engineering, illustrating the progression from in silico design to strain engineering and bioprocess optimization.
Diagram 2: Metabolic pathway for naringenin production in engineered E. coli, highlighting both native metabolism (gray) and heterologous enzymes (blue) introduced for biosynthesis [6].
The comparative analysis of TRY metrics across diverse biosynthetic pathways reveals several strategic implications for researchers and drug development professionals. First, the selection of microbial host should be guided not only by historical precedent but by systematic evaluation of the specific metabolic demands of the target pathway, as demonstrated by the superior naringenin production in S. cerevisiae versus E. coli [6]. Second, the integration of computational design tools like MCS analysis and DySScO with advanced gene editing technologies enables more predictable and effective pathway optimization [3] [2]. Third, the development of specialized fermentation strategies, such as two-stage pH control or cofactor feeding, can dramatically enhance TRY metrics even in extensively engineered strains [4].
As synthetic biology continues to expand the range of complex molecules accessible through microbial production, the strategic optimization of titer, yield, and productivity will remain essential for translating laboratory innovations into commercially viable bioprocesses. The frameworks, data, and methodologies presented in this guide provide a foundation for researchers to systematically approach this optimization challenge, balancing the inherent trade-offs between these critical metrics while advancing the frontier of sustainable chemical production.
Within industrial biotechnology, prolonged fermentation processes are critical for producing high-value biomolecules, from therapeutic proteins to alternative food ingredients. However, the productivity of these bioprocesses is intrinsically limited by the physiological decline of microbial and cellular workhorses. This review examines the critical limitations imposed by cellular aging and metabolic stress on prolonged fermentation, framing these challenges within the broader thesis of evaluating efficiency metrics for biosynthetic pathways. As living catalysts, the metabolic vitality of production organisms directly dictates the economic viability and scalability of fermentation-based manufacturing. A comparative analysis of experimental data reveals how aging-associated decline in metabolic function creates bottlenecks, providing a framework for researchers to quantify and overcome these barriers in pathway engineering and bioprocess optimization.
Cellular Hallmarks of Aging in Production Organisms
During extended fermentation, production organisms exhibit molecular and cellular changes that mirror hallmark aging processes, directly impacting metabolic output and culture longevity. These processes are conserved across model systems from yeast to mammalian cells.
- Genomic Instability and DNA Damage: Accumulation of DNA damage during prolonged culture activates DNA damage response (DDR) pathways, diverting cellular resources away from production and toward repair mechanisms. In yeast models, this damage is exacerbated by reactive oxygen species (ROS) generated as metabolic byproducts, particularly under industrial fermentation conditions [8] [9].
- Metabolic Dysregulation: Aging cells experience mitochondrial dysfunction and declining energy production. Integrated metabolic models of aging mouse gut microbiomes reveal a pronounced reduction in metabolic activity accompanied by downregulation of essential pathways in nucleotide metabolism critical for maintaining cellular replication and homeostasis during sustained fermentation [10].
- Loss of Proteostasis: With replicative age, cells progressively lose the ability to maintain protein homeostasis, leading to accumulation of misfolded proteins. This is particularly detrimental in precision fermentation where microbial hosts are engineered to overexpress recombinant proteins, creating substantial proteostatic stress that can trigger stress responses and reduce yields [9].
- Cellular Senescence: Production organisms can enter a state of irreversible growth arrest while remaining metabolically active but with altered secretion profiles. Senescent cells exhibit the senescence-associated secretory phenotype (SASP), releasing inflammatory cytokines and proteases in mammalian systems or altering metabolite secretion in microbial systems, which can negatively impact product quality and culture homogeneity [9].
Table 1: Hallmarks of Cellular Aging in Fermentation Systems
| Aging Hallmark | Impact on Fermentation Efficiency | Experimental Measurement |
|---|---|---|
| Genomic Instability | Reduced genetic fidelity, mutation accumulation | γ-H2AX foci, COMET assay [8] |
| Metabolic Dysregulation | Declining ATP production, reduced biosynthesis | NAD+/NADH ratio, ATP assays [10] |
| Loss of Proteostasis | Recombinant protein aggregation, reduced yields | Heat shock protein levels, aggregation assays [9] |
| Cellular Senescence | Culture growth arrest, altered product profile | β-galactosidase staining, SASP analysis [9] |
| Mitochondrial Dysfunction | Increased ROS, oxidative stress damage | ROS staining, mitochondrial membrane potential [8] |
Comparative Analysis of Aging Across Model Systems
Different production platforms exhibit distinct aging dynamics under industrial fermentation conditions. Understanding these system-specific aging trajectories is essential for selecting appropriate production hosts for long-duration bioprocesses.
Microbial Systems (Yeast/Bacteria)
The budding yeast Saccharomyces cerevisiae serves as a fundamental eukaryotic model for aging research due to its short lifespan and well-characterized genetics. Yeast aging studies have identified clear relationships between intracellular metabolites and aging under fermentation conditions. Specifically, trehalose levels increase with aging and under calorie restriction, indicating activation of protective responses against cellular stress during fermentation [11]. NMR-based metabolomics reveals that both calorie restriction and quercetin treatment significantly increase intracellular proline levels, which regulate mitochondrial function and decline with age, suggesting shared metabolic pathways for longevity promotion in fermentation environments [11].
Mammalian Cell Systems
Mammalian cells used in advanced fermentation applications exhibit more complex aging phenotypes. Primary cells have a finite replicative capacity—the Hayflick limit—before entering replicative senescence, fundamentally limiting their utility in prolonged bioprocesses [9]. Induced pluripotent stem cells (iPSCs) offer potential solutions but still retain aging signatures from donor cells. Research demonstrates that neurons from aged donors retain critical features of aging including reduced mitochondrial activity and increased ROS levels, which would directly impact their performance as production hosts in extended fermentations [9].
Table 2: System-Specific Aging Characteristics in Fermentation
| Production System | Key Aging Markers | Impact on Prolonged Fermentation |
|---|---|---|
| S. cerevisiae (Yeast) | Trehalose accumulation, proline decline, ROS increase [11] | Reduced ethanol tolerance, decreased recombinant protein yield |
| L. plantarum (Lactic Acid Bacteria) | Acid stress response, redox imbalance [12] | Reduced viability, altered metabolite profiles in fermented foods |
| Mammalian Cell Culture | Telomere attrition, SASP secretion, epigenetic alterations [9] | Growth arrest, altered product glycosylation, batch inconsistency |
| Filamentous Fungi | Hyphal fragmentation, autolysis [13] | Reduced enzyme secretion, morphology changes |
Metabolic Stress Pathways and Experimental Assessment
Metabolic stress during fermentation arises from intrinsic and extrinsic factors that collectively impact cellular aging and biosynthetic capacity. The interplay between these stressors and aging pathways creates a self-reinforcing cycle that accelerates functional decline in production organisms.
Diagram 1: Metabolic Stress Pathways in Prolonged Fermentation. Intrinsic and extrinsic stressors converge on core cellular damage pathways that ultimately impact fermentation performance.
Experimental Methodologies for Quantifying Aging and Stress
Research into fermentation-associated aging employs standardized assays to quantify both chronological and replicative lifespan under industrial conditions:
Chronological Lifespan (CLS) Assay: Measures the survival time of non-dividing cells in stationary phase, relevant for batch fermentation processes. Implementation involves spot assays where yeast cells are cultured in YPD media under different glucose concentrations (2.0%, 0.5%, 0.2% for calorie restriction studies), transferred to fresh media, and viability determined through serial dilution spotting on agar plates followed by incubation and colony counting [11].
Replicative Lifespan (RLS) Assay: Quantifies the number of daughter cells produced by a mother cell before senescence, critical for continuous fermentation systems. This typically uses biotin-streptavidin labeling or mother cell enrichment systems with micromanipulation to count progeny [11].
Metabolomic Profiling: ¹H nuclear magnetic resonance (NMR)-based metabolomics enables comprehensive quantification of intracellular metabolites during aging. Sample preparation involves adjusting cell densities to OD₆₀₀=20, washing pellets with phosphate buffer, quenching in liquid nitrogen, and metabolite extraction before analysis to identify aging signatures like trehalose and proline fluctuations [11].
Integrated Metabolic Modeling: Constraint-based reconstruction of metabolic networks from multi-omics data (metagenomics, transcriptomics, metabolomics) predicts metabolic fluxes and host-microbiome interactions during aging. This approach has revealed aging-associated declines in metabolic activity and reduced beneficial interactions in mouse gut microbiome studies, with applications to fermentation systems [10].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Studying Fermentation-Associated Aging
| Reagent/Category | Function in Aging Research | Specific Examples & Applications |
|---|---|---|
| Lifespan Assay Kits | Quantify replicative and chronological aging | Yeast CLS spot assay components [11] |
| Metabolic Probes | Detect mitochondrial function and ROS | H2DCFDA for ROS, TMRE for membrane potential [8] |
| Senescence Markers | Identify senescent cells in culture | β-galactosidase detection kits [9] |
| NMR Metabolomics | Comprehensive metabolite profiling | ¹H NMR instrumentation and protocols [11] |
| DNA Damage Assays | Quantify genomic instability | γ-H2AX antibodies, COMET assay kits [8] |
| Constraint-Based Modeling Tools | Predict metabolic flux changes | gapseq for metabolic network reconstruction [10] |
Cellular aging and metabolic stress represent fundamental bottlenecks in prolonged fermentation processes, directly impacting key efficiency metrics for biosynthetic pathways. The experimental data comparative analysis reveals that strategies targeting metabolic resilience—such as calorie restriction mimetics, antioxidant treatments, and proline supplementation—show promise in extending the productive lifespan of fermentation hosts. Future pathway engineering efforts should prioritize stability metrics alongside productivity, incorporating age-resilience as a design parameter in synthetic biology approaches. By quantifying and addressing these critical limitations, researchers can develop next-generation production systems that maintain metabolic vitality throughout prolonged fermentation cycles, ultimately enhancing the sustainability and economic viability of industrial biotechnology.
Comparative Analysis of Native vs. Heterologous Pathway Performance Metrics
In the development of microbial cell factories, a fundamental strategic choice involves utilizing a host's innate, native metabolic pathways versus introducing engineered, heterologous pathways from other organisms. This decision critically influences the overall efficiency, yield, and economic viability of bioproduction processes for chemicals, pharmaceuticals, and enzymes. Native pathways are integrated into the host's existing regulatory and metabolic networks, whereas heterologous pathways often provide a direct and optimized route to the target compound but require careful balancing with host physiology [14]. This guide provides an objective comparison of these approaches, underpinned by recent experimental data and performance metrics, to inform researchers and scientists in the field of drug development and metabolic engineering.
Performance Metrics Comparison
The performance of biosynthetic pathways is quantitatively assessed using three key metrics: titer (the concentration of the product, typically in mg/L or g/L), yield (the amount of product formed per unit of substrate, often in mol/mol or g/g), and productivity (the rate of product formation, in mg/L/h or g/L/h) [15]. The following tables summarize these metrics for various products from recent studies, comparing native and heterologous production routes.
Table 1: Performance Metrics for Metabolite Production in Engineered Strains
| Target Product | Host Organism | Pathway Type | Key Engineering Strategy | Max Titer (mg/L) | Yield (mol/mol glucose) | Productivity (mg/L/h) | Citation |
|---|---|---|---|---|---|---|---|
| Naringenin | E. coli | Heterologous | Step-wise enzyme screening & host engineering (M-PAR-121) | 765.9 (Shake-flask) | - | - | [6] |
| Pyridoxine (Vitamin B6) | Bacillus subtilis | Native & Heterologous | DXP-independent pathway & medium optimization | 174.6 (Fed-batch) | - | - | [16] |
| Indigoidine | E. coli BL21(DE3) | Heterologous | NRPS/PPTase screening & membrane engineering | 26,710 (Fed-batch) | - | - | [17] |
| L-Lysine | S. cerevisiae | Native (L-2-aminoadipate) | - | - | 0.8571 (YT) | - | [15] |
| L-Lysine | E. coli | Native (Diaminopimelate) | - | - | 0.7985 (YT) | - | [15] |
| L-Lysine | C. glutamicum | Native (Diaminopimelate) | - | - | 0.8098 (YT) | - | [15] |
Table 2: Performance Metrics for Heterologous Protein Production in Aspergillus niger [18]
| Target Protein | Origin | Expression Host | Engineering Strategy | Max Titer (mg/L) | Enzyme Activity |
|---|---|---|---|---|---|
| Glucose Oxidase (AnGoxM) | Aspergillus niger (Homologous) | A. niger AnN2 | TeGlaA copy reduction & PepA disruption | 416.8 | ~1276-1328 U/mL |
| Pectate Lyase (MtPlyA) | Myceliophthora thermophila | A. niger AnN2 | Site-specific integration & Cvc2 overexpression | 130.7 (+18%) | ~1627-2106 U/mL |
| Triose Phosphate Isomerase (TPI) | Bacterial | A. niger AnN2 | Site-specific integration | 110.8 | ~1751-1907 U/mg |
| Immunomodulatory Protein (LZ8) | Ganoderma lucidum | A. niger AnN2 | Site-specific integration | 163.3 | - |
Detailed Experimental Protocols
Protocol 1: De Novo Naringenin Production in E. coli
This protocol outlines the step-wise optimization of a heterologous pathway in E. coli for the high-titer production of naringenin, a plant polyphenol [6].
- Strain Construction: The heterologous pathway was constructed in the tyrosine-overproducing E. coli strain M-PAR-121. Genes were cloned into plasmid vectors (e.g., pRSFDuet-1, pCDFDuet-1) under inducible T7 promoters.
- Step-wise Pathway Validation:
- TAL Screening: Two tyrosine ammonia-lyase (TAL) genes from different sources were expressed in three E. coli strains (BL21(DE3), K-12 MG1655(DE3), M-PAR-121). Production of the intermediate p-coumaric acid was measured to select the best TAL (from Flavobacterium johnsoniae) and host (M-PAR-121) combination.
- 4CL and CHS Screening: The best TAL was combined with different 4-coumarate-CoA ligase (4CL) and chalcone synthase (CHS) genes. The combination of FjTAL, At4CL (Arabidopsis thaliana), and CmCHS (Cucurbita maxima) yielded the highest naringenin chalcone.
- CHI Screening: Different chalcone isomerase (CHI) genes were tested. CHI from Medicago sativa (MsCHI) was identified as the most effective for the final conversion to naringenin.
- Cultivation and Production: Production experiments were conducted in shake flasks. After initial growth, gene expression was induced, and cultures were supplemented with the carbon source. Naringenin production was quantified over time using HPLC after removing cell biomass via centrifugation.
Protocol 2: Platform for Heterologous Protein Expression in Aspergillus niger
This protocol describes the creation of a chassis strain and a modular platform for high-yield heterologous protein expression in the industrial fungus A. niger [18].
- Chassis Strain Development: The industrial glucoamylase hyperproducer A. niger AnN1 was engineered using a CRISPR/Cas9-assisted marker recycling system.
- Gene Copy Reduction: Thirteen of the 20 native tandem copies of the TeGlaA gene were deleted to reduce background protein secretion.
- Protease Disruption: The major extracellular protease gene PepA was disrupted to minimize degradation of the target heterologous protein. The resulting strain was named AnN2.
- Modular Protein Expression:
- Vector Construction: A donor DNA plasmid was designed with the native AAmy promoter and AnGlaA terminator as homologous arms for integration.
- Site-Specific Integration: Target genes (e.g., MtPlyA, LZ8) were integrated into the high-expression loci previously occupied by the deleted TeGlaA copies in the AnN2 chassis strain via CRISPR/Cas9.
- Secretory Pathway Engineering: To further enhance yield, the COPI vesicle trafficking component gene Cvc2 was overexpressed in strains expressing target proteins like MtPlyA.
- Cultivation and Analysis: Transformants were cultivated in shake flasks for 48-72 hours. Extracellular proteins in the culture supernatant were analyzed. Target protein titer was quantified, and enzyme activity was measured using specific activity assays.
Pathway and Workflow Visualization
The following diagrams illustrate the logical workflow for heterologous pathway optimization and the specific engineered pathways discussed in this guide.
Figure 1: A generalized workflow for the step-wise optimization of a heterologous biosynthetic pathway in a microbial host, as demonstrated for naringenin production [6].
Figure 2: The heterologous pathway for de novo naringenin production in E. coli. Enzyme abbreviations and their optimal sources identified in the study are: TAL (Tyrosine ammonia-lyase), 4CL (4-coumarate-CoA ligase), CHS (Chalcone synthase), CHI (Chalcone isomerase) [6].
The Scientist's Toolkit: Essential Research Reagents
This section details key reagents, strains, and molecular tools frequently employed in the construction and optimization of heterologous pathways.
Table 3: Key Research Reagents for Pathway Engineering
| Reagent / Tool | Function / Application | Specific Examples |
|---|---|---|
| Model Host Organisms | Microbial chassis for pathway integration and testing. | E. coli BL21(DE3), B. subtilis, S. cerevisiae, A. niger [15] [18] |
| Specialized Engineered Strains | Hosts with enhanced precursor supply for specific pathways. | E. coli M-PAR-121 (Tyrosine overproducer) [6] |
| Expression Vectors | Plasmids for cloning and expressing heterologous genes. | pRSFDuet-1, pCDFDuet-1, pACYCDuet-1 [6] |
| Genome Editing Systems | Tools for precise genomic modifications (deletions, integrations). | CRISPR/Cas9 system for A. niger [18] |
| Enzyme / Gene Libraries | Diverse sources of heterologous genes for pathway screening. | TAL, 4CL, CHS, CHI genes from various plants and microbes [6] |
| Computational Pathway Tools | Algorithms for in silico pathway design and host selection. | SubNetX for pathway extraction and ranking [19] |
| Genome-Scale Models (GEMs) | Metabolic models for predicting yield and flux analysis. | GEMs of E. coli, S. cerevisiae, etc., for calculating YT and YA [15] |
Within metabolic engineering and biosynthetic pathway research, the selection of an appropriate microbial host is a critical determinant of success. The model organisms Escherichia coli and Saccharomyces cerevisiae represent the two most extensively utilized platforms for the production of biofuels, pharmaceuticals, and commodity chemicals. Framed within a broader thesis on efficiency metrics for biosynthetic pathways, this guide provides an objective comparison of these organisms' inherent metabolic capabilities, supported by experimental data. Understanding their core physiological and genetic differences enables researchers to make informed, rational decisions for host selection to maximize titer, yield, and productivity for a given target compound [20].
Core Physiological and Metabolic Comparison
The fundamental divergence between the prokaryotic E. coli and the eukaryotic S. cerevisiae extends beyond cellular structure to their core metabolism, regulatory mechanisms, and tolerance to process conditions. These inherent characteristics directly influence their suitability for specific biosynthetic pathways.
Table 1: Core Physiological and Metabolic Characteristics
| Characteristic | Escherichia coli | Saccharomyces cerevisiae |
|---|---|---|
| Organism Type | Prokaryote (Bacterium) | Eukaryote (Yeast) |
| Metabolic Pathway | 1-deoxy-D-xylulose 5-phosphate (DXP) pathway [21] | Mevalonate (MVA) pathway [21] |
| IPP Precursors | Pyruvate & Glyceraldehyde-3-phosphate [21] | Acetyl-CoA [21] |
| Theoretical Max IPP Yield (Glucose) | Higher potential yield from glucose [21] | Lower potential yield from glucose due to carbon loss in Acetyl-CoA formation [21] |
| Preferred Carbon Sources | Wide range, including glycerol [22] | Sugars (e.g., glucose, sucrose) |
| Tolerance to Inhibitors | Can be engineered for high furfural tolerance [23] | Naturally high tolerance to low pH and osmotic pressure [21] |
| Post-Translational Modifications | Limited; inability to perform eukaryotic PTMs [24] | Extensive; capable of complex PTMs similar to higher eukaryotes [25] [24] |
| Cofactor Regeneration | Can be engineered for balanced NADPH/NADH usage [26] | Native strong tendency to regenerate NAD+ for anaerobic growth [27] |
| Subcellular Organization | Cytoplasmic production; can store hydrophobic products in enlarged membranes [26] | Compartmentalization; allows for harnessing organelles [21] |
| GRAS Status | Not classified as GRAS | Generally Regarded As Safe (GRAS) [27] [25] |
Quantitative Performance in Key Pathways
Direct comparative studies and organism-specific optimizations reveal performance disparities in the production of valuable compounds. The data below, drawn from peer-reviewed literature, highlights achievable titers and yields.
Table 2: Representative Production Metrics for Selected Compounds
| Product | Host | Titer | Yield | Key Engineering Strategy |
|---|---|---|---|---|
| Squalene | E. coli | 1267 mg/L [26] | N/R | Redox-balanced HMGR, membrane lipid remodeling, in situ extraction [26] |
| Lycopene | E. coli | N/R | N/R | Systematic computational search & gene deletion using MOMA [28] |
| Ethanol (from Crude Glycerol) | E. coli | ~2.5 g/L [22] | N/R | Microaerobic fermentation conditions [22] |
| S. cerevisiae | ~4.5 g/L [22] | N/R | Use of isolated or evolved strains [22] | |
| L-Threonine | E. coli | N/R | N/R | Model-driven parametric sensitivity analysis of key enzymes [28] |
| Artemisinic Acid | S. cerevisiae | 25 g/L [25] | N/R | Full pathway reconstruction & strain optimization [25] |
| Vinblastine | S. cerevisiae | N/R | N/R | Extensive genomic engineering (56 edits) [25] |
Experimental Protocols for Pathway Analysis and Engineering
In Silico Profiling of Terpenoid Production Potential
Objective: To computationally compare the theoretical potential of E. coli and S. cerevisiae for producing isopentenyl diphosphate (IPP), the universal terpenoid precursor [21].
Methodology:
- Network Reconstruction: Genome-scale metabolic models for both organisms are constructed. The model for E. coli typically incorporates 65 reactions and 50 metabolites, while the model for S. cerevisiae includes 69 reactions and 60 metabolites, focusing on central carbon metabolism [21].
- Stoichiometric Analysis: The carbon, energy, and redox stoichiometries of the native DXP (E. coli) and MVA (S. cerevisiae) pathways are analyzed independently of the host network.
- Elementary Mode Analysis (EMA): EMA is used to calculate all feasible steady-state flux distributions through the metabolic network. This identifies the theoretical maximum yield of IPP on a given carbon source (e.g., glucose, xylose, glycerol) without requiring kinetic parameters [21].
- Identification of Engineering Targets:
- Overexpression Targets: EMs are analyzed to pinpoint reactions in central metabolism whose overexpression could alleviate energy and redox deficiencies that limit terpenoid yield.
- Knockout Strategies: The concept of Constrained Minimal Cut Sets (cMCSs) is applied. This computational algorithm identifies a minimal set of gene deletions that obligately couple cell growth to a high yield of the desired product, forcing the organism to become a high-yielding factory [21].
Systems Metabolic Engineering for Squalene Production in E. coli
Objective: To enhance the production of the hydrophobic triterpene squalene in E. coli by addressing pathway efficiency and product storage [26].
Methodology:
- Cofactor Engineering:
- A hybrid 3-hydroxy-3-methyl glutaryl coenzyme A reductase (HMGR) system is developed by combining NADPH-dependent and NADH-preferred enzymes.
- This strategy balances the intracellular NADPH/NADH ratio, leading to increased precursor flux and a reported squalene titer of 852.06 mg/L [26].
- Membrane & Storage Engineering:
- To address the limited storage capacity for hydrophobic products, the membrane morphology is engineered.
- Overexpression of genes dgs, murG, and plsC generates lipid-enriched, elongated cells, creating more internal storage space. This intervention boosts squalene production to 970.86 mg/L [26].
- Process Optimization:
- A delayed induction strategy is implemented to separate the growth and production phases.
- An in situ recovery system using a 10% dodecane overlay is applied to continuously extract squalene from the culture, mitigating potential product toxicity. This final optimization achieves a final titer of 1267.01 mg/L in a 3 L bioreactor [26].
Metabolic Pathway Diagrams
The diagram illustrates the fundamental metabolic routes for producing the universal terpenoid precursors, IPP and DMAPP, in E. coli and S. cerevisiae. The DXP pathway in E. coli starts from the glycolysis intermediates glyceraldehyde-3-phosphate (GAP) and pyruvate (PYR). In contrast, the Mevalonate (MVA) pathway in S. cerevisiae initiates from acetyl-CoA (AcCoA). This divergence in precursor origin is a critical factor in the theoretical yield calculations, with the DXP pathway possessing a higher potential carbon yield from glucose [21].
This workflow provides a rational framework for selecting between E. coli and S. cerevisiae based on project-specific requirements and the metabolic characteristics of each organism. Key decision points include the complexity of the target molecule, the theoretical yield of the biosynthetic pathway, and the intended application of the final product [25] [21] [24].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents and Tools for Metabolic Engineering
| Reagent / Tool | Function | Application Example |
|---|---|---|
| CRISPR/Cas9 Systems | Enables precise genome editing, knockout, and insertion of heterologous pathways. [23] [25] | Used in S. cerevisiae for the complex engineering required to produce vinblastine (56 edits). [25] |
| Genome-Scale Metabolic Models (GEMs) | In silico models (e.g., iAF1260 for E. coli) that predict organism behavior and identify engineering targets. [28] | Used in Flux Balance Analysis (FBA) to predict gene knockout strategies for improving lycopene production. [28] |
| Constrained Minimal Cut Sets (cMCSs) | A computational algorithm to identify minimal gene knockouts that couple growth to high product yield. [21] | Identified knockout strategies for E. coli and S. cerevisiae to create IPP-overproducing strains. [21] |
| Heterologous Pathways | Introduction of non-native metabolic routes into a host chassis. | Introduction of the MVA pathway into E. coli to enhance terpenoid production, circumventing native regulation. [21] |
| Inducible Promoters (e.g., GAL, CUP1) | Tightly regulated promoters that control the timing and level of gene expression. [24] | Used in S. cerevisiae to control the expression of toxic proteins or to separate growth and production phases. [24] |
| In Situ Extraction Solvents (e.g., Dodecane) | An overlay solvent that continuously extracts hydrophobic products from the fermentation broth. [26] | Used in E. coli squalene production to reduce product toxicity and inhibition, boosting final titer. [26] |
A fundamental challenge in metabolic engineering is rewiring a microbe's core metabolism to channel carbon and energy toward a desired product, a process that often creates a metabolic burden and trade-off with cell growth [29]. The optimization of central precursor availability is therefore paramount. Platform strains with engineered central carbon metabolism (CCM)—encompassing glycolysis, the tricarboxylic acid (TCA) cycle, and the pentose phosphate pathway (PPP)—provide the foundational metabolic driving force, or flux, for diverse biosynthetic pathways [30]. This guide objectively compares the performance of major platform strain engineering strategies, providing the experimental data and methodologies essential for selecting the optimal chassis for a given biosynthetic goal.
Comparative Analysis of Platform Strain Performance
Different engineering strategies manipulate CCM to enhance the supply of key precursor metabolites. The table below summarizes the performance outcomes of several major approaches.
Table 1: Comparison of Platform Strain Engineering Strategies for Enhanced Metabolic Flux
| Engineering Strategy | Key Precursor Enhanced | Chassis Organism | Target Product | Reported Yield/Improvement | Key Experimental Data |
|---|---|---|---|---|---|
| Heterologous PHK Pathway [30] | Acetyl-CoA, E4P | S. cerevisiae | Fatty Acid Ethyl Esters | 5100 ± 509 g/CDW (cell dry weight) [30] | Overexpression of Adh2, Ald6, ACS; introduction of PHK pathway. |
| Heterologous PHK Pathway [30] | Acetyl-CoA, E4P | S. cerevisiae | p-Hydroxycinnamic Acid | 12.5 g/L (154.9 mg/g glucose yield) [30] | Promoter optimization & dynamic regulation post-PHK introduction. |
| Heterologous PDH Pathway [30] | Acetyl-CoA | S. cerevisiae | General Acetyl-CoA | ~2-fold increase in acetyl-CoA [30] | Expression of NADP+-dependent E. coli PDH pathway. |
| Dynamic Genetic Circuits [29] | Varies based on pathway | E. coli | Gamma-aminobutyric acid (GABA) | High-level production from glycerol [29] | Dynamic metabolic control circuit to balance growth and production. |
| Sensor-Driven Evolution [31] | Varies based on pathway | E. coli | Naringenin & Glucaric Acid | 36-fold and 22-fold increase, respectively [31] | Biosensor-coupled selection; 4 rounds of evolution. |
| Flux-Enhanced Cell Extracts [32] | Shikimate Pathway Precursors | E. coli Extract | Muconic Acid | 4.5 mg/L (enabled detection) [32] | Cell-free prototyping using extract from rewired strain. |
Experimental Protocols for Key Engineering Methodologies
Protocol: Engineering the Heterologous Phosphoketolase (PHK) Pathway
The introduction of the PHK pathway is a widely validated strategy to enhance acetyl-CoA and E4P supply [30].
- Gene Identification and Cloning: Identify and codon-optimize genes for phosphoketolase (PK) and phosphotransacetylase (PTA) from donor organisms (e.g., Aspergillus nidulans).
- Vector Construction: Clone the PK and PTA genes into an appropriate expression vector under the control of strong, constitutive, or inducible promoters.
- Strain Transformation: Introduce the constructed plasmid into the chassis organism (e.g., S. cerevisiae).
- Pathway Validation: Confirm functional enzyme expression via proteomics and measure the impact on central metabolism by tracking changes in intracellular metabolite pools (e.g., acetyl-CoA, E4P) using LC-MS.
- Host Strain Optimization (Optional): To further enhance flux, knock out competing pathways (e.g., phosphofructokinase in Yarrowia lipolytica to redirect glycolytic flux) or overexpress downstream pathway genes [30].
- Fermentation and Analysis: Perform fed-batch fermentation with glucose as a carbon source. Quantify product titer, yield, and productivity using HPLC or GC-MS.
Protocol: Sensor-Driven Evolution of Biosynthetic Pathways
This method uses biosensors to couple production of a target metabolite to cell fitness, enabling high-throughput evolution [31].
- Biosensor Selection/Engineering: Identify a natural transcriptional regulator or riboswitch responsive to the target chemical. Engineer its promoter to control the expression of a selectable marker gene (e.g., antibiotic resistance).
- Sensor-Selector Strain Construction: Integrate the biosensor circuit into the host genome. To minimize "cheater" cells, implement strategies like appending a degradation tag (ssrA tag) to the selector protein or mutating the Ribosome Binding Site (RBS) for fine-tuned translation [31].
- Library Generation: Use targeted genome-wide mutagenesis (e.g., MAGE) on genes predicted by Flux Balance Analysis (FBA) to be critical for the pathway. This creates a vast library of pathway variants.
- Toggled Selection Rounds:
- Positive Selection: Grow the mutant library under antibiotic pressure. Cells producing sufficient levels of the target metabolite will activate the biosensor and survive.
- Negative Selection: Counter-screen the enriched population under conditions where the selector gene is toxic without the inducer (e.g., using a different antibiotic or SDS). This eliminates cheaters that survive via sensor malfunction.
- Iteration and Analysis: Repeat steps 3-4 for multiple rounds. Isolate evolved strains and sequence their genomes to identify causative mutations. Validate production titers with analytical methods.
Visualizing Key Metabolic Engineering Concepts
Rewiring Central Carbon Metabolism with the PHK Pathway
The diagram below illustrates how the heterologous PHK pathway integrates into native CCM to enhance flux toward acetyl-CoA and E4P, key precursors for lipids and aromatics.
Biosensor-Driven Evolution Workflow
This flowchart outlines the iterative process of using a genetically encoded biosensor to evolve high-producing strains.
The Scientist's Toolkit: Essential Research Reagents and Solutions
This table details key genetic elements, strains, and methodologies that form the toolkit for flux enhancement research.
Table 2: Key Reagents and Resources for Flux Engineering Research
| Tool/Reagent | Category | Example/Description | Primary Function in Research |
|---|---|---|---|
| CRISPR-Cas Tools [33] | Genome Editing | CRISPR-based markerless mutagenesis in E. coli [33]. | Enables precise, scarless deletion of competing genes (e.g., waaL, wecA) and integration of pathway genes. |
| Genetic Circuits [29] | Dynamic Regulation | Circuits responsive to metabolic intermediates (e.g., malonyl-CoA, acetyl-CoA). | Automatically balances cell growth and product synthesis, preventing metabolic burden. |
| Biosensors [31] | Screening & Selection | Transcription factors (e.g., TtgR, TetR) or riboswitches coupled to reporter genes. | High-throughput screening of mutant libraries by linking metabolite concentration to fluorescence or survival. |
| Flux-Enhanced Strains [32] | Chassis Platform | E. coli and S. cerevisiae strains with rewired CCM (e.g., enhanced shikimate pathway flux). | Provides a pre-engineered background with high precursor supply for pathway prototyping. |
| Cell-Free Extracts [32] | Prototyping System | Lysates derived from metabolically rewired strains. | Allows for rapid in vitro testing of pathway enzymes and feasibility before in vivo implementation. |
| Flux Analysis Algorithms [34] | Computational Tool | Enhanced Flux Potential Analysis (eFPA). | Predicts relative metabolic flux changes by integrating proteomic or transcriptomic data at the pathway level. |
The data demonstrates that no single strategy is universally superior; the choice depends on the target product's metabolic demands. The heterologous PHK pathway is exceptionally powerful for products deriving from acetyl-CoA and E4P, such as fatty acids and aromatics [30]. In contrast, for pathways with complex regulation or unknown bottlenecks, sensor-driven evolution provides a powerful, non-rational method to explore a vast mutational landscape [31]. A prevailing trend is the move from static to dynamic regulation, where genetic circuits auto-regulate flux in response to metabolic status, thereby optimizing the growth-production trade-off [29].
Furthermore, the emergence of flux-enhanced strain toolkits and their corresponding cell-free extracts represents a paradigm shift, drastically accelerating the design-build-test-learn cycle [32]. Researchers can now prototype pathways in vitro using extracts with enhanced precursor supply, de-risking and informing subsequent in vivo engineering. When combined with advanced computational tools like eFPA that predict flux from omics data, these technologies provide an integrated, data-driven framework for engineering the next generation of microbial cell factories [34]. The ultimate efficiency metric in biosynthetic pathways research is the successful and rapid translation of a design into a strain that achieves industrially relevant titers, yields, and productivities, a goal now within closer reach thanks to these advanced platform strains and prototyping strategies.
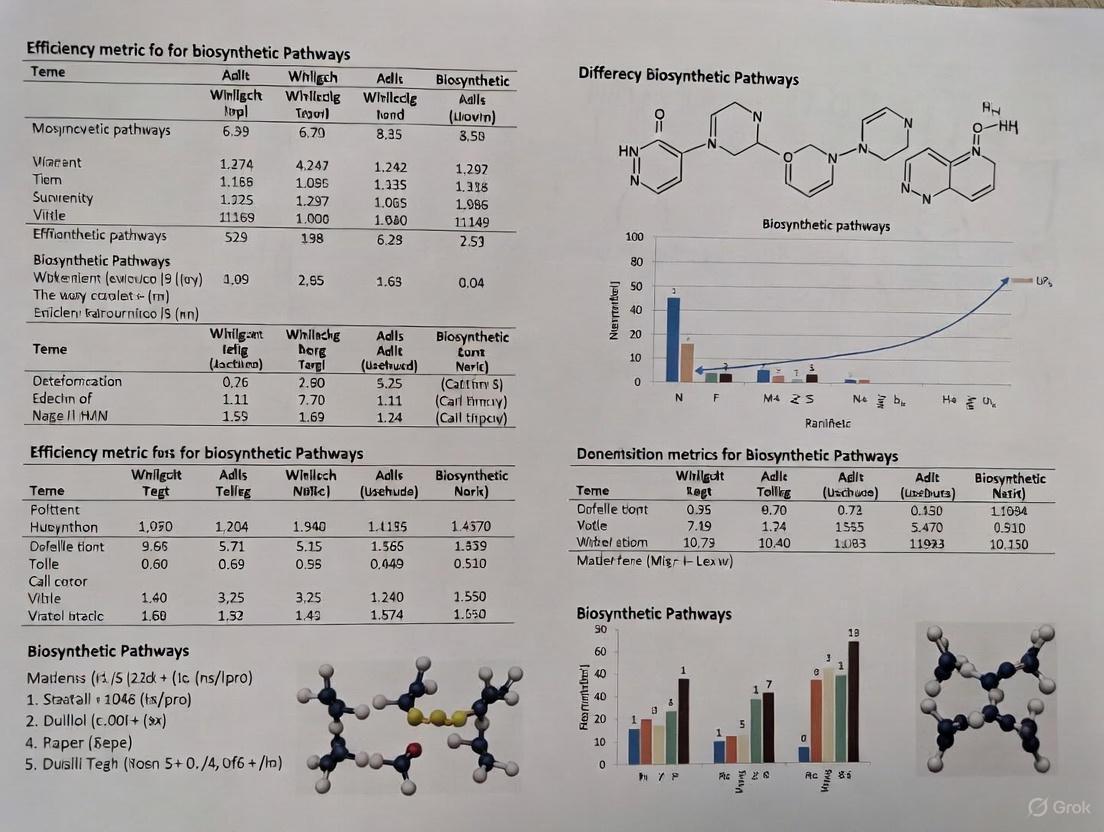
Computational and Experimental Methods for Pathway Design and Implementation
Retrosynthesis and enumeration algorithms are fundamental computational tools in metabolic engineering and synthetic biology. They enable the systematic design of biosynthetic pathways for the production of high-value compounds, from pharmaceuticals to industrial chemicals, by working backwards from a target molecule to identify feasible synthetic routes using available starting materials and enzymatic transformations. This guide provides an objective comparison of three prominent algorithms—FindPath, BNICE.ch, and RetroPath2.0—focusing on their operational principles, performance characteristics, and practical applications within a broader research context focused on efficiency metrics for biosynthetic pathways.
The following diagram illustrates the core operational workflows of BNICE.ch, RetroPath2.0, and FindPath, highlighting their distinct approaches to pathway exploration.
Performance and Application Comparison
The table below summarizes a direct comparison of the key operational and performance characteristics of BNICE.ch, RetroPath2.0, and FindPath, based on documented experimental implementations.
| Feature | BNICE.ch | RetroPath2.0 | FindPath |
|---|---|---|---|
| Core Approach | Generalized enzymatic reaction rules [35] | Retrosynthesis search from target to sink compounds [36] | Enumeration from host organism metabolism [36] |
| Primary Output | Network of all possible intermediates and pathways [35] | Specific retrosynthetic pathways leading to sink compounds [37] [36] | Biosynthetic pathways from a chassis organism's native metabolism [36] |
| Pathway Ranking | By popularity (citations/patents) and thermodynamic feasibility [35] | Not specified in results | By pathway length and Conserved Atom Ratio (CAR) [36] |
| Typical Application | Exploring chemical space for novel derivatives [35] | Finding feasible pathways to a target molecule [36] | Designing pathways within a specific chassis organism (e.g., E. coli) [36] |
| Experimental Validation | Used to discover pathways for (S)-tetrahydropalmatine and other BIA derivatives in yeast [35] | Integrated into workflows producing L-DOPA and dopamine in E. coli [36] | Integrated into workflows producing L-DOPA and dopamine in E. coli [36] |
| Reported Output (Sample) | Generated a network of 4,838 compounds and 17,597 reactions for noscapine pathway expansion [35] | Part of a workflow achieving 0.71 g/L L-DOPA and 0.29 g/L dopamine titers in E. coli [36] | Part of a workflow achieving 0.71 g/L L-DOPA and 0.29 g/L dopamine titers in E. coli [36] |
Detailed Experimental Protocols
Protocol for Pathway Expansion and Derivative Synthesis Using BNICE.ch
This protocol is adapted from research that expanded the noscapine biosynthetic pathway to produce analgesic and anxiolytic derivatives [35].
Workflow Diagram: BNICE.ch Pathway Expansion
Key Reagents and Solutions
- Software Tool: BNICE.ch with its library of generalized enzymatic reaction rules.
- Reference Database: Kyoto Encyclopedia of Genes and Genomes (KEGG) for validating known enzymatic functions.
- Host Organism: Engineered Saccharomyces cerevisiae (yeast) strains producing noscapine pathway intermediates.
- Target Compounds: (S)-tetrahydropalmatine and other benzylisoquinoline alkaloid (BIA) derivatives.
Protocol for Heterologous Pathway Implementation Using FindPath and RetroPath2.0
This protocol is adapted from a study that designed and implemented pathways in E. coli for the production of L-DOPA and dopamine [36].
Workflow Diagram: Integrated Pathway Design Workflow
Key Reagents and Solutions
- Software Suite: FindPath, BNICE.ch, RetroPath2.0, ShikiAtlas Retrotoolbox, BridgIT, Selenzyme.
- Host Organism: Escherichia coli (E. coli) production chassis.
- Analytical Technique: Ultra Performance Liquid Chromatography (UPLC) for quantifying target compound titers.
- Key Enzymes: Tyrosinase from Ralstonia solanacearum and DOPA decarboxylase from Pseudomonas putida for the L-DOPA-to-dopamine route.
The selection of an optimal retrosynthesis and enumeration algorithm is highly dependent on the specific research goals. BNICE.ch excels in the comprehensive exploration of chemical space to discover novel pathway derivatives. RetroPath2.0 is highly effective for finding feasible retrosynthetic routes from a target molecule to available building blocks. FindPath is optimal for designing pathways that are integrated into and extend the native metabolism of a specific chassis organism. As demonstrated in experimental workflows, these tools are often used in concert, leveraging their respective strengths to efficiently bridge the gap from computational design to successful in vivo implementation.
The construction of efficient biosynthetic pathways for producing value-added compounds is a central goal in synthetic biology. However, designing these pathways manually is challenging and time-consuming [38]. In recent years, computational workflows that integrate pathway generation algorithms with enzyme selection tools have emerged as powerful solutions. These platforms enable researchers to systematically design, evaluate, and implement biosynthetic routes for target molecules, significantly accelerating the development of microbial cell factories for pharmaceuticals, biofuels, and specialty chemicals.
This guide provides an objective comparison of integrated computational frameworks for biosynthetic pathway design, focusing on their core methodologies, performance characteristics, and experimental validation. The analysis is framed within a broader research context of developing efficiency metrics for biosynthetic pathways, providing drug development professionals and researchers with critical insights for tool selection and implementation.
Comparative Analysis of Integrated Platforms
The table below summarizes the core capabilities and experimental validation of major integrated platforms for computer-aided pathway design.
Table 1: Comparison of Integrated Computational Platforms for Biosynthetic Pathway Design
| Platform Name | Primary Approach | Pathway Design Tools | Thermodynamic Assessment | Enzyme Selection Method | Experimental Validation |
|---|---|---|---|---|---|
| novoStoic2.0 [39] | Stoichiometry-based pathway synthesis with thermodynamic evaluation | novoStoic, optStoic | dGPredictor | EnzRank (CNN-based scoring) | Hydroxytyrosol pathways (shorter routes, reduced cofactor usage) |
| Computational Workflow [40] | Retrosynthesis and enumeration with structure-based gene discovery | FindPath, BNICE.ch, RetroPath2.0 | N/A | GDEE pipeline (homology modeling & docking) | L-DOPA (0.71 g/L) and dopamine (0.21-0.29 g/L) production in E. coli |
| COMPSS Framework [41] | Generative protein sequence evaluation with composite metrics | N/A (focuses on enzyme evaluation) | N/A | Composite metrics (alignment-based, alignment-free, structure-based) | Malate dehydrogenase & copper superoxide dismutase (70-90% identity to natural) |
| BNICE.ch Workflow [35] | Biochemical network expansion and enzyme prediction | BNICE.ch | N/A | BridgIT | (S)-tetrahydropalmatine production in yeast |
Experimental Protocols and Performance Data
Pathway Implementation for Tyrosine-Derived Compounds
Experimental Protocol: Researchers developed a computational workflow integrating retrosynthesis algorithms (FindPath, BNICE.ch, RetroPath2.0) with a structure-based gene discovery pipeline (GDEE) for selecting enzymes [40]. The methodology involved:
- Pathway Generation: Using ShikiAtlas Retrotoolbox to enumerate pathways from tyrosine to L-DOPA and dopamine with maximum 30 reaction steps and minimum atom conservation ratio of 0.34.
- Enzyme Selection: Applying the GDEE pipeline utilizing homology modeling with Modeller and molecular docking with AutoDock Vina to rank candidate enzymes based on binding affinity as a proxy for catalytic efficiency.
- Implementation: Cloning selected gene candidates into E. coli for shake-flask experiments using a mutant tyrosinase from Ralstonia solanacearum for L-DOPA production and DOPA decarboxylase from Pseudomonas putida for dopamine production.
Performance Data: The implemented pathways achieved a maximum L-DOPA titer of 0.71 g/L and dopamine titers of 0.29 g/L (known pathway) and 0.21 g/L (novel pathway) [40]. This demonstrated the workflow's effectiveness in identifying functional biosynthetic routes, including the first validated alternative pathway for dopamine in microbes.
Computational Scoring of Generated Enzymes
Experimental Protocol: A comprehensive evaluation of computational metrics for predicting enzyme functionality was conducted over multiple experimental rounds [41]:
- Sequence Generation: Three generative models (ESM-MSA, ProteinGAN, and Ancestral Sequence Reconstruction) produced sequences for malate dehydrogenase (MDH) and copper superoxide dismutase (CuSOD).
- Metric Evaluation: Twenty diverse computational metrics were assessed, including alignment-based (sequence identity), alignment-free (language model likelihoods), and structure-based scores (AlphaFold2 confidence).
- Experimental Testing: Over 500 natural and generated sequences with 70-90% identity to natural sequences were expressed, purified, and assayed for in vitro activity.
Performance Data: Initial "naive" generation resulted in mostly inactive sequences (only 19% of tested sequences were active) [41]. However, Ancestral Sequence Reconstruction showed notably better performance, generating 9/18 active CuSOD enzymes and 10/18 active MDH enzymes. The developed COMPSS computational filter improved the rate of experimental success by 50-150% compared to unfiltered approaches.
Workflow Visualization
The following diagram illustrates the logical flow and component integration in a comprehensive computer-aided workflow for biosynthetic pathway design, from initial target specification to experimental implementation.
Table 2: Key Research Reagents and Computational Tools for Pathway Engineering
| Tool/Reagent | Function/Purpose | Example Applications |
|---|---|---|
| RetroPath2.0 [40] | Retrosynthesis workflow for pathway design | Enumeration of novel pathways from starting compounds to targets |
| BNICE.ch [40] [35] | Biochemical Network Integrated Computational Explorer for pathway expansion | Generation of hypothetical chemical space around pathway intermediates |
| Selenzyme [40] | Enzyme selection tool for suggested reactions | Recommendation of appropriate gene candidates for desired transformations |
| BridgIT [40] [35] | Enzyme-reaction matching through structural similarity | Identification of enzymes for novel reactions by similarity to known reactions |
| dGPredictor [39] | Thermodynamic feasibility assessment | Estimation of standard Gibbs energy changes for novel reactions |
| EnzRank [39] | CNN-based enzyme-substrate compatibility scoring | Rank-ordering known enzymes for novel substrate activity |
| AutoDock Vina [40] | Molecular docking for binding affinity prediction | Ranking candidate enzymes in structure-based gene discovery pipelines |
| E. coli BL21(DE3) [40] [42] | Heterologous expression host for pathway implementation | Production of L-DOPA, dopamine, and other target compounds |
Integrated computational platforms have significantly advanced the field of biosynthetic pathway design by combining multiple tools into cohesive workflows. The comparison reveals distinct strengths across platforms: novoStoic2.0 provides comprehensive thermodynamic evaluation, the GDEE workflow [40] demonstrates robust experimental validation with measurable product titers, and the COMPSS framework [41] offers sophisticated enzyme functionality prediction. These tools collectively enable researchers to navigate the complex journey from pathway conception to experimental implementation with increasing predictive accuracy and success rates.
For drug development professionals, these integrated approaches offer promising strategies for accelerating the production of pharmaceutical compounds and their derivatives, ultimately contributing to more efficient and sustainable biomanufacturing pipelines. As these platforms continue to evolve, they will likely incorporate more sophisticated machine learning approaches and expanded biochemical databases to further improve their predictive capabilities and experimental success rates.
Natural Products (NPs) are organic compounds synthesized by living organisms and represent a vital source for drug discovery, with over 60% of FDA-approved small-molecule drugs being NPs or their derivatives [43] [44]. However, the biosynthetic pathways for over 90% of natural products remain uncharacterized, creating a major bottleneck for their scalable production and engineering [44]. Traditional rule-based computational methods face significant challenges in predicting these complex pathways.
Deep learning approaches are overcoming these limitations by enabling template-free retrosynthetic analysis. This guide provides an objective performance comparison of BioNavi-NP, a dedicated toolkit for NP biosynthetic pathway prediction, against other emerging computational tools, with experimental data contextualized within efficiency metrics for biosynthetic pathway research.
Performance Comparison of Deep Learning Tools for Biosynthetic Pathway Prediction
Extensive benchmarking studies reveal how different computational tools perform on standardized datasets, allowing researchers to select the most appropriate solution for their specific needs. The table below summarizes the key performance metrics of leading tools.
Table 1: Performance Comparison of BioNavi-NP and Alternative Tools on Standard Benchmarks
| Tool / Model | Core Approach | Single-Step Top-1 Accuracy (%) | Single-Step Top-10 Accuracy (%) | Multi-Step Pathway Recovery Rate (%) | Key Differentiating Features |
|---|---|---|---|---|---|
| BioNavi-NP [43] | Transformer + AND-OR Tree Search | 21.7 (Ensemble) | 60.6 (Ensemble) | 72.8 | Data augmentation with organic reactions; Navigable AND-OR tree planning |
| GSETransformer [44] | Graph-Sequence Enhanced Transformer | Information not available in search results | State-of-the-art on BioChem benchmarks | Information not available in search results | Integrates molecular graph data with SMILES sequences |
| READRetro [44] | Ensemble (Graph2SMILES + Retroformer) | Information not available in search results | Competitive results on BioChem benchmarks | Information not available in search results | Ensemble model combining graph and sequence-based architectures |
| RetroPathRL [43] | Rule-based + Reinforcement Learning | ~10.6 (Estimated from comparison) | ~42.1 (Estimated from comparison) | Information not available in search results | Conventional rule-based approach; Lower accuracy than deep learning methods |
BioNavi-NP demonstrates a significant performance advantage, with its top-10 single-step accuracy being 1.7 times higher than conventional rule-based approaches like RetroPathRL [43]. Furthermore, it successfully identified biosynthetic pathways for 90.2% of test compounds and recovered the exact reported building blocks for 72.8% of them in multi-step planning tests [43]. The emerging GSETransformer model highlights a trend toward integrating structural graph information with sequential SMILES data to better handle molecular complexity [44].
Experimental Protocols and Workflows
Understanding the experimental methodologies used to generate performance data is crucial for interpreting results and planning new research.
BioNavi-NP's Training and Evaluation Protocol
BioNavi-NP's performance was validated through a rigorously defined experimental workflow [43].
- Dataset Curation (BioChem): The model was primarily trained on a dataset curated from public databases like MetaCyc, KEGG, and MetaNetX, containing 33,710 unique precursor-metabolite pairs [43].
- Data Augmentation (USPTO_NPL): To enhance robustness, the training set was expanded with 62,370 organic reactions involving natural product-like compounds from the USPTO database, creating a combined dataset of ~96,000 reactions [43].
- Model Architecture and Training: The core single-step prediction model uses a Transformer neural network, an attention-based architecture effective for sequence-to-sequence tasks. An ensemble of four such models was employed to improve prediction robustness [43].
- Multi-Step Planning Algorithm: For multi-step pathways, BioNavi-NP employs a deep learning-guided AND-OR tree-based search algorithm. This efficiently navigates the combinatorial explosion of possible routes by strategically expanding the most promising precursor candidates [43].
- Evaluation Metrics: The model was evaluated on a held-out test set of 1,000 biosynthetic reactions for single-step accuracy and 368 internal test cases for multi-step pathway recovery [43].
The following diagram visualizes this integrated workflow for biosynthetic pathway prediction.
Figure 1: BioNavi-NP's integrated workflow combines data from biological and chemical sources with a two-stage prediction process.
Benchmarking Protocol for Comparative Studies
Independent studies comparing multiple tools, such as the evaluation of GSETransformer, follow a standardized protocol to ensure fairness [44].
- Benchmark Datasets: Models are trained and tested on public benchmarks like USPTO-50K (for general organic synthesis) and BioChem Plus (for biosynthesis). The dataset is split into training, validation, and test subsets (e.g., 80%/10%/10%) [44].
- Strict Splitting: To evaluate generalization, a "clean" dataset version is sometimes created by removing all reactions present in the multi-step test set from the training data, preventing data leakage [44].
- Consistent Evaluation Metrics: All models are compared using the same metrics, primarily top-k accuracy for single-step prediction and pathway recovery rate for multi-step planning [44].
Essential Research Reagent Solutions for Computational Pathway Prediction
The development and application of tools like BioNavi-NP rely on a foundation of publicly available data and software resources. The table below catalogues key reagents for computational biosynthetic research.
Table 2: Key Research Reagents and Databases for Computational Biosynthesis
| Resource Name | Type | Primary Function in Research | Relevance to Pathway Prediction |
|---|---|---|---|
| KEGG [45] [46] | Reaction/Pathway Database | Reference repository of known metabolic pathways and enzymes. | Source of known pathways for training and validation; reference for pathway reconstruction. |
| MetaCyc [43] [46] | Reaction/Pathway Database | Curated database of experimentally elucidated metabolic pathways and enzymes. | Provides high-quality, curated biochemical reactions for model training. |
| USPTO [43] | Reaction Database | Large repository of organic chemical reactions extracted from patents. | Source for data augmentation to improve model robustness and generalizability. |
| PubChem [46] | Compound Database | Public repository of chemical compound structures and properties. | Essential for compound look-up, structure verification, and property calculation. |
| BRENDA [46] | Enzyme Database | Comprehensive enzyme information database detailing function and kinetics. | Used for linking predicted biochemical reactions to plausible enzymes. |
| RXNMapper [44] | Software Tool | Automated atom-mapping tool for chemical reactions. | Critical pre-processing step to define reaction centers in training data for template-free models. |
| Selenzyme / E-zyme [43] | Software Tool | Enzyme prediction tools that recommend potential enzymes for a given reaction. | Downstream application to assign putative enzymes to each step in a predicted pathway. |
The logical relationship between these resources in a typical research pipeline is illustrated below.
Figure 2: Research reagent workflow shows how data flows from foundational databases through analysis tools to final predictions.
Deep learning approaches like BioNavi-NP represent a significant advancement over traditional rule-based systems for predicting the biosynthetic pathways of natural products. Quantitative benchmarks demonstrate its superior accuracy in single-step retrosynthesis and high efficacy in multi-step pathway recovery.
The field is rapidly evolving, with new architectures like GSETransformer pushing the boundaries of performance by more effectively integrating molecular structure information. For researchers in drug discovery and metabolic engineering, these tools are becoming indispensable for accelerating the elucidation of complex biosynthetic pathways, thereby facilitating the sustainable production of valuable plant natural products and novel bioactive compounds [47]. The continued integration of large-scale multi-omics data with sophisticated deep learning models promises to further unlock the synthetic potential of natural product biosynthesis.
Multi-omics integration represents a transformative approach in biological research, enabling a holistic interpretation of molecular intricacy across multiple levels including genome, transcriptome, and metabolome [48]. This paradigm has revolutionized the field of medicine and biology by creating avenues for integrated system-level approaches that bridge the gap from genotype to phenotype [48]. For researchers investigating biosynthetic pathways, multi-omics provides powerful tools to unravel the complex interplay between genes, their expression patterns, and the resulting metabolic outputs that define cellular functions. Integrated approaches combine individual omics data, either sequentially or simultaneously, to understand the interplay of molecules and assess the flow of information from one omics level to another [48]. The advent of high-throughput techniques and availability of multi-omics data generated from large sample sets has catalyzed the development of numerous computational tools and methods for data integration and interpretation, creating new opportunities for discovering genes involved in specialized metabolism [48] [49].
For biosynthetic pathway research, efficiency metrics are increasingly dependent on multi-omics approaches that can simultaneously capture genomic potential, transcriptional activity, and metabolic outputs. Where single-omics studies provide limited snapshots of biological systems, integrated multi-omics enables researchers to connect genetic blueprints with functional outcomes, thereby accelerating the identification of key genes and regulatory elements controlling biosynthetic pathways [49]. This comprehensive review examines current methodologies, performance comparisons, and practical implementations of multi-omics integration specifically for gene discovery in biosynthetic pathways, providing researchers with critical insights for selecting appropriate strategies based on their specific research objectives and available data types.
Performance Comparison of Multi-omics Integration Methods
Benchmarking Results Across Methodologies
Multi-omics integration methods demonstrate varying performance characteristics depending on data types, biological context, and analytical goals. The table below summarizes quantitative performance metrics for prominent integration approaches applied to biosynthetic pathway discovery and gene identification.
Table 1: Performance Comparison of Multi-omics Integration Methods
| Method | Omics Layers Integrated | Primary Application | Reported Accuracy/Performance | Key Strengths |
|---|---|---|---|---|
| BioNavi-NP [43] | Genomic, Metabolomic | Natural product biosynthetic pathway prediction | 90.2% pathway identification rate; 72.8% building block recovery (1.7x better than rule-based) | Deep learning-based; handles complex natural products |
| MINIE [50] | Transcriptomic, Metabolomic | Causal network inference | Significant improvement over state-of-art methods; robust performance in curated networks | Bayesian approach; handles timescale separation; infers causal relationships |
| Network Propagation [51] | Genomic, Transcriptomic, Metabolomic | Drug target identification | Varies by implementation; superior for identifying novel disease modules | Leverages prior biological knowledge; captures system-level properties |
| Graph Neural Networks [51] | Multi-omics layers | Drug response prediction | High accuracy in heterogeneous data integration | Captures complex non-linear relationships; adaptable to various network structures |
| Early Data Fusion (Concatenation) [52] | Genomic, Transcriptomic, Metabolomic | Genomic prediction | Inconsistent results; often underperforms vs. model-based integration | Simple implementation; minimal preprocessing requirements |
| Model-based Fusion [52] | Genomic, Transcriptomic, Metabolomic | Complex trait prediction | Consistently improves predictive accuracy over genomic-only models | Captures non-additive, nonlinear, and hierarchical interactions |
Performance Analysis and Method Selection Guidelines
The benchmarking data reveals that method performance significantly depends on the specific research objective. For biosynthetic pathway elucidation, deep learning approaches like BioNavi-NP demonstrate superior performance in identifying complete pathways and recovering known building blocks [43]. Transformer neural networks trained on both biochemical and organic reactions achieve top-10 precursor prediction accuracy of 60.6%, substantially outperforming conventional rule-based approaches [43].
For inferring regulatory mechanisms and causal relationships, Bayesian methods like MINIE that explicitly model temporal dynamics and timescale separation between molecular layers show significant advantages [50]. These approaches successfully capture the reality that metabolic processes occur on much faster timescales (minute-level) compared to transcriptional changes (hour-level), leading to more biologically plausible network inferences [50].
In genomic prediction contexts, model-based integration strategies consistently outperform simple data concatenation approaches, particularly for complex traits influenced by multiple biological layers [52]. Methods that capture non-additive, nonlinear, and hierarchical interactions across omics layers provide more accurate predictions of phenotypic outcomes, enabling more efficient selection in breeding programs [52].
Experimental Protocols for Multi-omics Integration
Protocol 1: Integrated Pathway Discovery Using BioNavi-NP
Objective: Identification of complete biosynthetic pathways for natural products using multi-omics data.
Experimental Workflow:
Data Preparation: Curate genomic and metabolomic data for target organism. For novel natural products, obtain high-resolution mass spectrometry data and NMR spectra for structural elucidation [43].
Single-step Retrosynthesis Prediction:
- Input target natural product as SMILES representation
- Apply ensemble transformer neural networks to predict potential biosynthetic precursors
- Generate top-k precursor candidates (typically k=10) based on trained model [43]
Multi-step Pathway Planning:
- Implement AND-OR tree-based search algorithm to explore combinatorial pathway options
- Iterate single-step predictions recursively until reaching known building blocks
- Rank complete pathways by computational cost, length, and organism-specific enzyme availability [43]
Experimental Validation:
- Express candidate genes in heterologous system (e.g., yeast, E. coli)
- Analyze metabolic intermediates using LC-MS/MS
- Confirm pathway completeness through isotope labeling experiments
Figure 1: BioNavi-NP Pathway Discovery Workflow
Protocol 2: Causal Network Inference with MINIE
Objective: Infer regulatory networks integrating transcriptomic and metabolomic data to identify key regulatory genes.
Experimental Workflow:
Time-Series Data Collection:
- Collect single-cell RNA-seq data at multiple time points (e.g., 0, 6, 12, 24, 48 hours)
- Obtain bulk metabolomic measurements from same biological system at matched time points [50]
Data Preprocessing:
- Normalize transcript counts using standard scRNA-seq pipelines
- Transform metabolomic data using probabilistic quotient normalization
- Align temporal measurements across omics layers
Network Inference:
- Implement differential-algebraic equation model to handle timescale separation
- Apply Bayesian regression framework to infer network topology
- Incorporate curated metabolic reaction networks as prior knowledge [50]
Validation and Interpretation:
- Perform gene ontology enrichment on identified regulatory genes
- Validate key interactions through targeted gene knockdown experiments
- Compare network topology with known pathway databases
Figure 2: MINIE Causal Network Inference Protocol
Computational Tools and Databases
Table 2: Essential Multi-omics Research Resources
| Resource | Type | Function | Application in Biosynthetic Pathways |
|---|---|---|---|
| TCGA [48] | Data Repository | Provides multi-omics data for cancer samples | Comparative analysis of secondary metabolism in disease contexts |
| ICGC [48] | Data Repository | Coordinates large-scale cancer genome studies | Access to somatic mutation data affecting metabolic pathways |
| CCLE [48] | Data Repository | Gene expression, copy number, sequencing from cancer cell lines | Screening model systems for pathway engineering |
| METABRIC [48] | Data Repository | Clinical traits, expression, SNP, and CNV from breast tumors | Understanding metabolic adaptations in disease |
| plantiSMASH [49] | Analysis Tool | Identifies biosynthetic gene clusters in plants | Discovery of novel specialized metabolic pathways |
| BioNavi-NP [43] | Analysis Tool | Predicts biosynthetic pathways for natural products | De novo pathway design and reconstruction |
| MINIE [50] | Analysis Tool | Infers multi-omic networks from time-series data | Identifying regulatory genes controlling metabolic fluxes |
| Selenzyme [43] | Analysis Tool | Selects plausible enzymes for predicted reactions | Enzyme assignment in putative biosynthetic pathways |
| E-zyme 2 [43] | Analysis Tool | Predicts enzyme commission numbers | Functional annotation of pathway genes |
| HyperGCN [53] | Analysis Tool | Integrative analysis of spatial transcriptomics | Spatial localization of pathway expression |
Experimental Reagents and Platforms
Sequencing Technologies: Single-cell RNA sequencing platforms (10x Genomics, Drop-seq) enable transcriptome profiling at cellular resolution, crucial for understanding heterogeneous biosynthetic systems [50]. Bulk RNA-seq remains valuable for overall pathway expression analysis.
Mass Spectrometry Platforms: High-resolution LC-MS/MS systems (Orbitrap, Q-TOF) provide sensitive detection and quantification of metabolites, enabling comprehensive metabolomic profiling of biosynthetic pathways [54].
CRISPR-Cas9 Systems: Genome editing tools facilitate functional validation of identified genes through targeted knockout and knockdown experiments in native or heterologous hosts.
Heterologous Expression Systems: Model microbial hosts (S. cerevisiae, E. coli) and plant systems (N. benthamiana) enable functional characterization of putative biosynthetic pathways identified through multi-omics integration [49] [43].
Multi-omics integration represents a powerful paradigm for gene discovery in biosynthetic pathway research, with method selection critically dependent on specific research goals. For novel pathway elucidation, deep learning approaches like BioNavi-NP demonstrate superior performance in navigating the complex chemical space of natural products [43]. For understanding regulatory mechanisms, causal inference methods like MINIE that explicitly model biological timescales provide more accurate insights into gene-metabolite relationships [50]. In agricultural contexts, model-based integration of genomic, transcriptomic, and metabolomic data significantly enhances prediction accuracy for complex traits [52].
The future of multi-omics integration in biosynthetic research will likely involve increased incorporation of spatial context [53], three-dimensional tissue modeling [53], and dynamic temporal resolution [50]. As methods continue to evolve, researchers must balance computational sophistication with biological interpretability to ensure that multi-omics insights can be effectively translated into practical applications in drug discovery, crop improvement, and industrial biotechnology. By strategically selecting integration methods aligned with specific research objectives and leveraging the growing toolkit of multi-omics resources, scientists can significantly accelerate the pace of gene discovery and pathway engineering across diverse biological systems.
Differential expression (DE) analysis serves as a fundamental methodology in computational biology for identifying genes that show significant expression changes across different biological conditions, tissues, or experimental treatments. In biosynthetic pathway research, DE analysis provides the critical first step for pinpointing key enzymatic genes and regulatory elements that drive metabolic flux toward desired natural products. The integration of DE findings with pathway enrichment analysis allows researchers to move from individual gene lists to comprehensive biological insights, revealing how entire metabolic networks respond to genetic, environmental, or developmental perturbations. For pharmaceutical and biotechnology applications, this approach accelerates the identification of rate-limiting steps in valuable compound synthesis and informs metabolic engineering strategies. This guide objectively compares the performance of leading DE methodologies and visualization tools, evaluating their applicability across various research scenarios in pathway-focused investigations.
Comparative Performance of Differential Expression Methods
Table 1: Benchmarking of Differential Expression Tools Across Data Types
| Method | Data Type | Statistical Approach | Key Strengths | Limitations | Recommended Use Cases |
|---|---|---|---|---|---|
| DESeq2 [55] | Bulk RNA-seq | Negative binomial generalized linear model with empirical Bayes shrinkage | High specificity, robust dispersion estimation, handles low counts well | Cannot model random effects, requires adequate biological replicates | Standard bulk RNA-seq experiments with standard experimental designs |
| DREAM [55] | Bulk or Pseudobulk RNA-seq | Linear mixed model with empirical Bayes shrinkage on VST-transformed data | Accounts for repeated measures and complex correlations | Longer computation time than standard linear models | Studies with paired samples, repeated measurements, or atlas-level data |
| MAST [55] | Single-cell RNA-seq | Generalized linear mixed effects hurdle model | Explicitly models the bimodality of single-cell data (zero inflation) | Computationally intensive, requires VST-transformed data | Single-cell DE analysis where accounting for technical zeros is critical |
| Permutation Test [55] | Pseudobulk RNA-seq | Non-parametric resampling | Distribution-free, minimal assumptions | Computationally expensive, minimum p-value limitation ( \frac{1}{\text{permutations}} ) | Small sample sizes or when data distribution assumptions are violated |
| Hierarchical Bootstrapping [55] | Single-cell RNA-seq | Non-parametric resampling with hierarchical structure | Properly accounts for pseudoreplication in nested designs | Newer method with less established benchmarks | Single-cell data with clear hierarchical structure (cells within samples) |
The benchmarking of these methods reveals that pseudobulk approaches generally outperform methods designed specifically for single-cell data when applied to individual datasets, with DESeq2 showing particularly robust performance in standardized bulk RNA-seq experiments [55]. For atlas-level analyses or studies with complex correlations, DREAM provides an optimal balance between analytical performance and computational efficiency. A critical consideration across all methods is the proper accounting for biological replication, as treating individual cells as independent observations dramatically inflates type I error rates due to pseudoreplication bias [55].
Experimental Protocols for Differential Expression Analysis
Standard Bulk RNA-seq Analysis Pipeline
Protocol 1: Identification of Differentially Expressed Genes from Bulk RNA-seq Data
*Sample Preparation and Sequencing*: Extract high-quality RNA from biological replicates (minimum n=3 per condition). Prepare libraries using standardized kits (e.g., Illumina TruSeq) and sequence on an appropriate platform to achieve minimum depth of 20-30 million reads per sample.
*Quality Control and Alignment*: Assess raw read quality using FastQC. Trim adapters and low-quality bases with Trimmomatic or Cutadapt. Align cleaned reads to a reference genome using splice-aware aligners such as STAR or HISAT2.
*Read Quantification*: Generate count matrices for genes or transcripts using featureCounts or HTSeq, ensuring proper handling of multimapping reads and ambiguity.
*Normalization and Differential Expression*: Import count matrices into R/Bioconductor. Normalize for library size and composition biases using methods inherent to DESeq2 (median-of-ratios) or edgeR (TMM). Perform statistical testing for differential expression using appropriate design matrices that account for experimental factors.
*Multiple Testing Correction*: Apply false discovery rate (FDR) control using the Benjamini-Hochberg procedure. Consider a threshold of padj < 0.05 and |log2FC| > 1 as statistically significant for most applications.
*Pathway Integration*: Input significant DEGs into enrichment tools such as Reactome or DAVID for functional interpretation and pathway analysis [56].
Tissue-Specific Expression Analysis Protocol
Protocol 2: Cross-Tissue Expression Profiling for Pathway Gene Identification
*Data Collection*: Access large-scale transcriptomic datasets such as GTEx (6,665 samples across 25 tissues) or tissue-specific expression compendia [57].
*Tissue-Specific Expression Quantification*: Calculate reads per kilobase per million (RPKM) or transcripts per million (TPM) to enable cross-sample comparison. Establish expression thresholds (e.g., RPKM ≥ 1) to define actively expressed genes in each tissue [57].
*Specificity Assessment*: Classify genes as ubiquitously expressed (≥20 tissues), specifically expressed (≤5 tissues), or intermediate. Note that disease-associated genes are disproportionately represented among ubiquitously expressed genes (Odds Ratio 2.08) [57].
*Affected Tissue Mapping*: Integrate with disease phenotype databases such as OMIM and Human Phenotype Ontology (HPO) to associate genes with affected tissues [57].
*Expression-Phenotype Correlation*: Use receiver-operating characteristics (ROC) analysis to evaluate associations between elevated expression in specific tissues and phenotypic manifestations (maximum AUC = 0.69) [57].
Multi-Condition Experimental Design
Protocol 3: Analyzing Transcriptome Reprogramming Across Multiple Conditions
*Experimental Design*: Expose model organisms (e.g., C57BL/6J mice) to systematically varied environmental conditions (e.g., 10°C, 22°C, 34°C for thermal adaptation studies) with appropriate acclimation periods [58].
*Multi-Tissue Sampling*: Collect a comprehensive panel of tissues relevant to the biological process (e.g., for thermal adaptation: spleen, bone marrow, spinal cord, brain, hypothalamus, ileum, liver, quadriceps, and multiple adipose depots) [58].
*Batch-Robust Processing*: Process all RNA samples simultaneously using standardized library preparation to minimize technical variation. Utilize randomized block designs in sequencing runs.
*Tissue-Specific Differential Expression*: Perform DE analysis separately for each tissue type while maintaining consistent statistical thresholds. For the thermal adaptation study, this revealed adipose tissues underwent the most severe transcriptome changes, followed by immune tissues and the central nervous system [58].
*Cross-Tissue Meta-Analysis*: Identify conserved response genes versus tissue-specific regulators. In thermal adaptation, despite common stimuli, transcriptional responses exhibited a high degree of tissue-specificity at both the gene and gene ontology enrichment levels [58].
Visualization Approaches for Differential Expression Data
Table 2: Visualization Tools for Interpreting Differential Expression Results
| Tool | Visualization Type | Key Features | Data Compatibility | Integration Capabilities |
|---|---|---|---|---|
| Pathway Volcano [59] | Interactive volcano plots filtered by pathways | Reactome API integration, pathway-focused filtering, interactive inspection | Output from DESeq2, edgeR, or other DE tools | Direct connection to Reactome pathway database |
| bigPint [60] | Parallel coordinate plots, scatterplot matrices | Interactive clustering visualization, outlier detection, multi-gene patterns | RNA-seq count data and DE results | Standalone R package with minimal dependencies |
| Standard Volcano Plot | Static volcano plot | Log2FC vs. -log10(p-value) representation, highlight significant genes | Any DE results with fold changes and p-values | Compatible with standard graphing tools like ggplot2 |
Effective visualization is essential for interpreting complex differential expression datasets. The standard volcano plot provides a foundational approach for visualizing the relationship between statistical significance (p-value) and biological effect size (fold change). However, emerging tools like Pathway Volcano address the challenge of overplotting in traditional volcano plots by enabling pathway-focused filtering, allowing researchers to distill thousands of genes into biologically meaningful patterns [59]. Similarly, bigPint offers interactive capabilities for identifying clustered patterns of expression across multiple samples, facilitating the detection of both consistent and anomalous expression profiles [60].
Figure 1: Differential Expression Analysis Workflow. This diagram outlines the standard pipeline from experimental design through validation.
Table 3: Key Research Reagent Solutions for Differential Expression Studies
| Reagent/Resource | Category | Function | Example Applications |
|---|---|---|---|
| GTEx Dataset [57] | Reference Database | 6,665 tissue-wide transcriptomes across 25 human tissues | Establishing baseline tissue-specific expression patterns |
| Reactome [59] | Pathway Knowledgebase | Curated pathway database with API access | Pathway enrichment analysis and visualization |
| DESeq2 [55] | Statistical Software | Differential expression analysis of count-based RNA-seq data | Identifying significantly regulated genes between conditions |
| OMIM/HPO [57] | Phenotype Database | Catalog of human genes and genetic disorders with phenotype associations | Linking expression patterns to disease manifestations |
| R/Bioconductor | Computational Environment | Open-source statistical programming platform | Implementing DE analysis pipelines and custom visualizations |
| Illumina RNA-seq Kits | Library Preparation | Preparation of sequencing libraries from RNA samples | Generating transcriptome data for DE analysis |
| MetaCyc/KEGG [43] | Metabolic Pathway Database | Curated biosynthetic pathways and enzyme functions | Placing DEGs in metabolic context for pathway engineering |
The comparative evaluation of differential expression methodologies reveals that optimal tool selection depends critically on experimental design, data structure, and research objectives. For standard bulk RNA-seq analyses, DESeq2 remains the gold standard due to its robust statistical framework and extensive community validation. In studies with complex random effects or repeated measures, DREAM provides enhanced modeling flexibility without sacrificing performance. For single-cell applications, pseudobulk approaches consistently outperform methods designed specifically for single-cell data when applied to individual datasets, though hierarchical bootstrapping emerges as a promising approach for properly accounting for nested structures [55].
The integration of DE analysis with pathway-focused visualization tools like Pathway Volcano creates a powerful framework for moving from gene lists to biological insight [59]. Furthermore, the systematic assessment of expression patterns across tissues—as demonstrated in the GTEx and thermal adaptation studies—provides critical context for interpreting the functional significance of differentially expressed genes in pathway analysis [57] [58]. As biosynthetic pathway research increasingly focuses on multi-tissue and multi-condition responses, these integrated approaches for differential expression analysis will remain essential for identifying key regulatory genes and prioritizing targets for metabolic engineering and therapeutic development.
Advanced Strategies for Overcoming Bottlenecks and Enhancing Production
In the pursuit of constructing efficient microbial cell factories for chemical production, a significant challenge emerges: cellular aging and metabolic stress during prolonged fermentation processes drastically reduce productivity. Conventional metabolic engineering focuses on optimizing pathway flux, but this approach often neglects cellular robustness and longevity. Emerging research establishes chronological lifespan (CLS) extension as a critical efficiency metric in biosynthetic pathway performance, creating a paradigm where cellular longevity and biosynthetic capacity are fundamentally interconnected [61] [62].
This guide examines a transformative strategy that integrates lifespan engineering with traditional metabolic engineering. The core hypothesis is that extending the productive lifespan of microbial factories through targeted genetic interventions automatically remodels cellular metabolism to enhance overall robustness. We will objectively compare the performance of this approach against conventional methods, focusing on quantitative data from peer-reviewed studies and providing the experimental protocols necessary for implementation [61].
Core Mechanisms and Rationale
The Nutrient Sensing-Mitophagy Axis in Cellular Aging
The lifespan engineering strategy rests on two pivotal cellular processes: nutrient sensing and mitophagy.
- Nutrient Sensing Pathways: Signaling pathways such as Target of Rapamycin (TOR) and Protein Kinase A (PKA) act as the cell's internal fuel gauges. In nutrient-rich environments, these pathways are active and promote processes like protein synthesis and cell growth, but they simultaneously suppress stress resistance and longevity. Weakening nutrient sensing mimics a caloric restriction state, shifting the cell's focus from proliferation to maintenance and survival, thereby extending its functional lifespan [61] [63].
- Mitophagy: This is a selective form of autophagy that removes damaged or dysfunctional mitochondria. As a quality control mechanism, enhancing mitophagy prevents the accumulation of reactive oxygen species (ROS) and cellular damage, which are key drivers of aging. It is regulated by pathways like PINK1-Parkin, where PINK1 accumulates on damaged mitochondria and recruits the E3 ubiquitin ligase Parkin, initiating the engulfment of mitochondria by autophagosomes [64].
The rational combination is synergistic. Weakening nutrient signaling reduces anabolic pressure and induces a pro-survival state, while enhanced mitophagy ensures mitochondrial fitness within that state. Omics data reveals that this combination remodels central metabolism and upregulates the expression of lifespan-related genes, creating a more robust cellular chassis for production [61] [62].
Visualizing the Integrated Signaling Pathway
The following diagram illustrates the core signaling pathways involved in this lifespan engineering strategy and their logical interactions.
Performance Data and Comparative Analysis
Quantitative Comparison of Biosynthetic Output
The most compelling evidence for the lifespan engineering approach comes from direct comparison of production metrics. The following table summarizes the quantitative performance of this strategy against a baseline engineered strain for the production of sclareol, a valuable diterpenoid.
Table 1: Performance Comparison of Lifespan Engineering vs. Conventional Metabolic Engineering for Sclareol Production in Yeast [61] [62]
| Engineering Strategy | Sclareol Titer (g/L) | Yield (g/g Glucose) | Percentage Increase in Titer | Key Genetic Modifications |
|---|---|---|---|---|
| Baseline High-Producing Strain | 11.8 | 0.027 | - | Metabolic pathway optimization only |
| + Weakened Nutrient Sensing | 16.5 | 0.038 | +39.8% | e.g., Deletion of SCH9 |
| + Enhanced Mitophagy | 15.1 | 0.035 | +28.0% | e.g., Overexpression of ATG32 |
| Combined Strategy | 20.1 | 0.046 | +70.3% | Weakened nutrient sensing & enhanced mitophagy |
| Combined Strategy + Central Metabolism Enhancement | 25.9 | 0.051 | +119.5% | Full integrated approach |
The data demonstrates a clear synergistic effect. The combination of nutrient sensing weakening and enhanced mitophagy resulted in a 70.3% increase in sclareol titer, significantly greater than the individual modifications. This synergy underscores that the two processes act on complementary biological mechanisms to boost cellular performance. The final integrated approach, which further optimized central metabolism, achieved the highest reported sclareol production in microbes at 25.9 g/L [61] [62].
Generalizability Across Product Classes
A key strength of an engineering strategy is its broad applicability. Research indicates that lifespan engineering is not product-specific. The same combination of weakening nutrient sensing and enhancing mitophagy also improved the biosynthesis of other valuable compounds, including [61] [62]:
- Sesquiterpenes: β-Elemene
- Phenolic Acids
This demonstrates the generalizability of the approach for developing robust microbial cell factories across different metabolic pathways.
Experimental Protocols
Workflow for Implementing Lifespan Engineering
A typical experimental workflow for implementing and validating this strategy in a yeast cell factory involves the following key stages, from genetic construction to system-level analysis.
Detailed Methodologies
1. Strain Construction in S. cerevisiae [61]
- Gene Deletion/Weakening: To weaken nutrient sensing, delete or downregulate key genes like SCH9 (a homolog of mammalian Akt/S6K) or components of the RAS-PKA pathway using CRISPR-Cas9 or homologous recombination.
- Gene Overexpression: To enhance mitophagy, overexpress core mitophagy genes such as ATG32 (the mitophagy receptor in yeast) or ATG11 under a strong constitutive promoter.
- Combined Strain: Create a strain harboring both the nutrient sensing modification and the mitophagy enhancement.
2. Chronological Lifespan (CLS) Assay [61]
- Culture: Grow yeast strains to stationary phase in synthetic complete medium.
- Incubation: Maintain cultures at 30°C with mild shaking.
- Viability Monitoring: At regular intervals (e.g., every 2-3 days), take aliquots and measure:
- Colony Forming Units (CFUs): Serially dilute and spot on YPD plates to determine the number of viable cells.
- Survival Rate: Calculate the percentage of viable cells relative to day 2 (set as 100% survival).
- Data Analysis: Plot survival percentage over time. An extended CLS is indicated by a slower death rate and a higher percentage of surviving cells at later time points.
3. Fed-Batch Fermentation & Metabolite Analysis [61] [62]
- Fermentation: Perform controlled fed-batch fermentations in bioreactors to provide a consistent nutrient supply and maintain optimal physiological conditions over an extended period.
- Metabolite Extraction: Extract organic compounds from the culture broth at defined time points using solvent extraction (e.g., with ethyl acetate or methanol).
- Product Quantification: Analyze samples using techniques like Gas Chromatography (GC) or Liquid Chromatography-Mass Spectrometry (LC-MS/MS). Quantify the target product (e.g., sclareol) by comparing against a standard curve of a purified standard.
The Scientist's Toolkit: Key Research Reagents
The following table lists essential reagents, materials, and tools required to implement the described lifespan engineering strategy.
Table 2: Essential Research Reagents and Tools for Lifespan Engineering
| Category | Item / Model Organism | Specific Example / Strain | Function / Application |
|---|---|---|---|
| Model Organism | Saccharomyces cerevisiae | BY4741 (common background) | Eukaryotic microbial cell factory chassis |
| Genetic Tools | CRISPR-Cas9 System | Plasmid sets for gene editing | Targeted gene deletion/insertion |
| Overexpression Vectors | pRS42X series with strong promoters (e.g., pTDH3) | Constitutive high-level gene expression | |
| Key Genetic Targets | Nutrient Sensing Genes | SCH9, RAS2, TPK1 | Weakening nutrient signaling pathways |
| Mitophagy Genes | ATG32, ATG11 | Enhancing mitochondrial clearance | |
| Analytical Instruments | Bioreactor System | DASGIP, BioFlo | Controlled fed-batch fermentation |
| LC-MS/MS System | e.g., Agilent 6470 series | Accurate quantification of target metabolites | |
| GC-FID/MS System | e.g., Agilent 8890 GC | Quantification of volatile products (e.g., terpenes) | |
| Culture Consumables | Synthetic Defined Medium | SD -Ura (for plasmid maintenance) | Selective growth of engineered strains |
| YPD Plates | Standard recipe | For CFU counting during CLS assays |
The integration of lifespan engineering, specifically the combination of weakened nutrient sensing and enhanced mitophagy, represents a paradigm shift in metabolic engineering. Moving beyond the optimization of isolated pathways to engineer the holistic physiology of the microbial host results in unprecedented gains in both product titer and yield, as demonstrated by the >100% increase in sclareol production. This strategy enhances cellular robustness and biosynthetic stability during the critical late stages of fermentation, addressing a fundamental bottleneck in industrial bioprocesses. By providing a generalizable framework for building more resilient cell factories, lifespan engineering establishes a new efficiency metric for biosynthetic pathway research—one that prioritizes longevity and stability alongside flux.
The optimization of biosynthetic pathways for chemical production is a central challenge in metabolic engineering and synthetic biology. A significant bottleneck in this process is the high-throughput screening of microbial variants to identify rare, high-producing cells. Sensor-selector systems have emerged as a powerful solution, using transcription factor-based biosensors to link the intracellular concentration of a target metabolite to a selectable or screenable reporter gene [65]. This enables the direct enrichment of high-producing cells from large libraries. However, the practical application of these systems is often hampered by the phenomenon of "cheater" cell enrichment, where low-producers are falsely selected due to cross-feeding of the target molecule [66]. This guide compares the performance of different evolution-guided optimization strategies that integrate sensor-selector systems, evaluating their efficacy in mitigating cheater enrichment and enriching for genuine high-producers, with a focus on efficiency metrics for biosynthetic pathway research.
Comparative Analysis of Sensor-Guided Optimization Platforms
The table below summarizes the performance and characteristics of different sensor-guided optimization approaches as applied to specific metabolic engineering goals.
Table 1: Performance Comparison of Sensor-Guided Optimization Platforms
| Optimization Platform / Strategy | Target Molecule(s) | Key Performance Metrics | Reported Fold-Increase in Production | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|
| Evolution-Guided Optimization [65] | Naringenin, Glucaric Acid | Final titer, Production fold-increase | Naringenin: 36-fold, Glucaric Acid: 22-fold | General strategy; combines mutagenesis with artificial selection; handles large libraries (~10^9 cells) | Requires a specific sensor for each target; potential for cheater enrichment |
| Biosensor Desensitization [66] | trans-Cinnamic Acid (tCA) | kcat improvement of PAL enzyme, Cheater suppression | kcat: ~70% increase (after single sort) | Effective cheater suppression via Carbon Catabolite Repression (CCR) | Requires optimization of biosensor response (e.g., via media conditions) |
| Evolution-Guided Bayesian Optimization (EGBO) [67] | Silver Nanoparticles (Optical properties, reaction rate, seed usage) | Hypervolume, PF coverage, Feasible solution rate | N/A (Solves multi-objective problems) | Excellent for constrained multi-objective optimization; good PF coverage | Developed for material synthesis parameters; less direct for pathway engineering |
| Biosensor with Orthogonal Pre-screen [66] | trans-Cinnamic Acid (tCA) | Cheater population reduction, kcat improvement | kcat: ~70% increase (after single sort) | Combats cheaters effectively by pre-screening true negatives | Adds a step to the screening workflow, increasing complexity |
Table 2: Quantitative Outcomes of Directed Evolution Campaigns
| Enzyme/Pathway Target | Sensor Type | Number of Evolution Rounds | Key Identified Mutations | Final Titer/Activity | Reference |
|---|---|---|---|---|---|
| Naringenin Pathway | Sensor for naringenin or precursor | 4 | Untargeted mutations from whole-genome sequencing | 61 mg/L from glucose | [65] |
| Phenylalanine Ammonia-Lyase (PAL) | HcaR-based biosensor for tCA | 1 (with desensitization & pre-screen) | Not specified | ~70% higher kcat | [66] |
| Glucaric Acid Pathway | Sensor for glucaric acid | 4 | Not specified | 22-fold increase from baseline | [65] |
Experimental Protocols for Key Methodologies
Core Evolution-Guided Optimization Workflow
The foundational protocol for evolution-guided optimization involves iterative cycles of diversification and selection [65].
- Library Generation: Create a diverse library of pathway variants. This can be achieved through targeted genome-wide mutagenesis of genes identified by flux balance analysis as critical to the pathway.
- Sensor-Selector Coupling: Engineer a sensor circuit where the intracellular target chemical controls the expression of a reporter gene necessary for survival under selective conditions (e.g., an antibiotic resistance gene).
- Artificial Selection: Subject the large library (addressing ~10^9 cells per round) to selective conditions. Cells that produce sufficient amounts of the target chemical will activate the sensor, express the survival gene, and proliferate.
- Cheater Suppression: Implement a negative selection scheme between rounds to eliminate "cheater" cells that survive without producing the target metabolite, thereby preserving library diversity.
- Iteration: Repeat the process for multiple rounds (e.g., up to four rounds) to progressively enrich for high-producing variants.
- Validation: Sequence the genomes of evolved strains to identify beneficial mutations, which can reveal new, untargeted routes for further optimization.
Protocol for Biosensor Desensitization to Suppress Cheaters
This protocol details a method to reduce false-positive enrichment when using a biosensor for enzyme engineering [66].
- Biosensor Construction: Clone a transcription factor (e.g., HcaR for tCA) and its corresponding promoter driving a reporter gene (e.g., sfGFP) into a plasmid.
- Mock Library Preparation: Create a co-culture of true positive cells (e.g., expressing a functional PAL enzyme) and true negative cells (e.g., with a non-functional PAL, tagged with a constitutively expressed BFP).
- Desensitization via Media: Grow the mock library in media containing a repressing carbon source like glucose. Carbon Catabolite Repression (CCR) naturally desensitizes the biosensor, raising its limit of detection (LOD) and activation threshold.
- Validation of Desensitization: Measure the biosensor's response (e.g., fluorescence) to exogenously added target metabolite in repressing (glucose) and non-repressing (glycerol) media. A higher EC50 in repressing media confirms desensitization.
- FACS with Orthogonal Pre-screen: Before the main FACS sort, use the constitutive tag (e.g., BFP) to pre-screen and gate out a large fraction of the true negative population, further reducing the potential cheater carryover.
- Isolation and Characterization: Sort the remaining population and isolate individual clones for characterization of enzyme activity (e.g., kcat) or product titer.
EGBO for Constrained Multi-Objective Optimization
This protocol outlines the EGBO algorithm for problems with multiple, conflicting objectives [67].
- Problem Formulation: Define the decision variables (e.g., reactant flow rates) and the multiple objectives to be optimized (e.g., spectral signature match, reaction rate, minimal seed usage). Specify any constraints (e.g., to prevent clogging).
- Initial Sampling: Perform an initial design of experiments (DoE) to sample the decision space.
- Parallel Optimization: In each iteration: a. Bayesian Optimization (qNEHVI): Use a Bayesian optimizer to propose candidate solutions that maximize the hypervolume improvement, balancing exploration and exploitation. b. Evolutionary Algorithm Selection: Apply selection pressure from an evolutionary algorithm to the population, guiding the search towards the Pareto Front (PF).
- Evaluation: Synthesize and characterize the proposed candidates using the high-throughput experimental platform (e.g., microfluidic droplet generator with hyperspectral imaging).
- Model Update: Update the surrogate models for the objectives and constraints with the new experimental data.
- Iteration: Repeat steps 3-5 until the evaluation budget is exhausted or a satisfactory PF is achieved.
Visualizing Workflows and Signaling Pathways
Biosensor Mechanism and Cheater Crosstalk
Diagram 1: Biosensor Crosstalk
Evolution-Guided Optimization Cycle
Diagram 2: EGO Cycle
Cheater Suppression Strategy
Diagram 3: Cheater Suppression
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Sensor-Selector System Development
| Reagent / Tool Category | Specific Example(s) | Function in Experiment |
|---|---|---|
| Transcription Factor Biosensors | HcaR-based sensor for trans-cinnamic acid [66]; Sensors for naringenin, glucaric acid [65] | Core detection element; binds target metabolite and activates reporter gene expression. |
| Reporter Genes | Fluorescent Proteins (sfGFP, BFP) [66]; Antibiotic resistance genes [65] | Provides a screenable (FACS) or selectable (survival) output linked to metabolite concentration. |
| Model Host Organisms | Escherichia coli MG1655 [66]; Engineered E. coli for flavonoid production [65] | Standard microbial chassis for pathway engineering and biosensor implementation. |
| Library Generation Tools | Targeted genome-wide mutagenesis [65]; CRISPR-Cas9 [68] | Creates genetic diversity in pathway genes or regulatory elements for evolution. |
| High-Throughput Screening | Fluorescence-Activated Cell Sorting (FACS) [66]; Microdroplet-based screening [66] | Enables physical isolation of high-producing cells from large libraries (>10^9). |
| Analytical Validation | HPLC [66]; Whole-genome sequencing [65] | Validates production titers and identifies beneficial mutations after enrichment. |
| Desensitization Reagents | Carbon sources for CCR (e.g., Glucose) [66] | Media components used to tune biosensor sensitivity and suppress cheater activation. |
In the development of microbial cell factories, a fundamental challenge persists: the inherent trade-off between cell growth and product synthesis. Engineered microbial strains often face metabolic stress during long-term fed-batch fermentation, leading to diminished fitness and reduced productivity in critical late-stage production phases [69] [61]. Chronological lifespan (CLS), defined as the time nondividing cells remain viable in stationary phase, has emerged as a crucial factor for industrial bioprocessing efficiency. Recent research demonstrates that targeted extension of CLS in yeast (Saccharomyces cerevisiae) significantly enhances production of valuable compounds including diterpenoids, sesquiterpenes, and phenolic acids [61]. This guide systematically compares the performance of three primary CLS extension strategies—nutrient sensing modulation, stress resistance enhancement, and central metabolism engineering—providing researchers with experimental data and methodologies for implementing these approaches in biosynthetic pathway optimization.
Comparative Analysis of CLS Extension Strategies
Table 1: Performance Comparison of CLS Extension Strategies for Bioproduction
| Strategy | Key Mechanisms | Model System | Production Improvement | CLS Extension | Key Experimental Evidence |
|---|---|---|---|---|---|
| Calorie Restriction | Reduced glucose (2%→0.5%); Enhanced mitobiogenesis; Reduced mtDNA mutations | Yeast (BY4743 strain) | N/A (lifespan focus) | Significant extension (p < 0.05) | Increased autophagosome formation; Reduced cell death [70] |
| TOR Pathway Modulation | Decreased TOR signaling; Enhanced stress resistance; Nuclear relocalization of Msn2 | Yeast deletion strains | N/A (lifespan focus) | Increased stationary phase survival | Pharmacological inhibition (rapamycin); Amino acid restriction [71] |
| Integrated Longevity Engineering | Weakened nutrient-sensing; Enhanced mitophagy; Central metabolism enhancement | Engineered yeast | Sclareol: +70.3% (20.1 g/L); Optimized: 25.9 g/L (0.051 g/g glucose) | Significant extension demonstrated | Upregulation of lifespan-related genes; Metabolic remodeling [61] |
| Metabolite Supplementation | Proline accumulation; Trehalose increase; Oxidative stress reduction | Yeast (D452-2 strain) | N/A (lifespan focus) | Significant extension via shared pathway with CR | NMR-based metabolomics; 1H NMR analysis [11] |
Table 2: Quantitative Production Outcomes from CLS Engineering
| Target Compound | Host System | Base Production | Enhanced Production | Yield Improvement | Strategy Employed |
|---|---|---|---|---|---|
| Sclareol | Engineered yeast | 11.8 g/L | 20.1 g/L | +70.3% | Weakened nutrient-sensing + enhanced mitophagy [61] |
| Sclareol (optimized) | Engineered yeast | 11.8 g/L | 25.9 g/L | +119.5% | Additional central metabolism enhancement [61] |
| β-elemene | Engineered yeast | Not specified | Significant improvement | Reported | CLS engineering strategy [61] |
| Phenolic acids | Engineered yeast | Not specified | Significant improvement | Reported | CLS engineering strategy [61] |
Experimental Protocols for CLS Assessment and Engineering
Chronological Lifespan Assay Protocol
The CLS assay is performed via spot assays according to an established protocol with modifications [11]. Key steps include:
Pre-culture Conditions: Yeast cells are seeded in YPD media (1.0% yeast extract, 2.0% peptone, and 2.0% glucose) and incubated at 300 rpm and 30°C for 24 hours.
Main Culture: Cells are washed and transferred to fresh SC media under controlled glucose conditions (typically 0.2%, 0.5%, or 2.0% glucose concentration) and incubated for an additional 72 hours. These cultured yeast cells are designated "day 0" for aging studies.
Viability Assessment: Aliquots of culture are serially diluted tenfold in sterile water and spotted onto YPD agar plates. Plates are incubated for 2 days, and viability is determined by colony formation.
Quantification: CLS is measured as the duration cells maintain viability in the stationary phase, with assessments typically performed at regular intervals over several days or weeks.
Calorie Restriction Implementation
Calorie restriction is implemented using YPD media containing reduced glucose concentrations (0.5% or 0.2% instead of standard 2.0%) [70] [11]. This simple dietary intervention significantly extends chronological lifespan through multiple mechanisms including enhanced mitochondrial function and reduced oxidative damage.
Integrated Longevity Engineering Workflow
Diagram 1: Integrated Longevity Engineering Workflow. This strategy combines multiple interventions to automatically remodel cellular metabolism for enhanced robustness and production [61].
Signaling Pathways in CLS Extension
Diagram 2: Signaling Pathways in Chronological Lifespan Extension. Multiple intervention points can extend CLS through different but potentially complementary mechanisms [11] [71].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for CLS and Bioproduction Studies
| Reagent/Condition | Function/Application | Example Usage | Key References |
|---|---|---|---|
| Reduced Glucose Media | Implements calorie restriction; extends CLS | YPD with 0.5% or 0.2% glucose instead of 2% | [70] [11] |
| Rapamycin | TOR pathway inhibition; pharmacological CLS extension | Added to culture media at specified concentrations | [71] |
| Quercetin | Antioxidant treatment; reduces oxidative stress | 0.05-0.2 mg/50 mL DMSO; extends CLS via AMPK | [11] |
| S. cerevisiae Strain D452-2 | Eukaryotic model for aging studies | CLS assays with defined genetic background | [11] |
| S. cerevisiae BY4743 | Model for genetic studies of aging | Glucose restriction effects on mtDNA and autophagy | [70] |
| 1H NMR Spectroscopy | Metabolomic analysis; identifies longevity biomarkers | Quantifies trehalose, proline changes in CR | [11] |
| YPD Media | Standard growth medium for yeast studies | Base medium for CLS assays and genetic studies | [70] [11] |
| Synthetic Complete (SC) Media | Defined medium for controlled experiments | Growth assays and precise nutritional manipulation | [11] |
Discussion: Implications for Biosynthetic Pathway Efficiency
The experimental evidence demonstrates that chronological lifespan extension directly enhances bioproduction efficiency, particularly for compounds requiring extended fermentation periods. The most impressive results come from integrated approaches that combine multiple longevity strategies rather than single-pathway interventions [61]. The 70.3% improvement in sclareol production (increasing to 119.5% with central metabolism enhancement) achieved through synergistic engineering of nutrient-sensing pathways and mitophagy represents a significant advance in microbial cell factory development [61].
From the perspective of efficiency metrics for biosynthetic pathways, CLS extension addresses the fundamental trade-off between growth and production by enhancing cellular robustness during the non-growth production phase. This approach aligns with strategies to balance cell growth and product synthesis through orthogonal design, dynamic regulation, and fermentation process control [69]. The automatic metabolic remodeling observed in CLS-extended strains [61] suggests that longevity engineering may provide a more sustainable solution to metabolic burden than direct pathway optimization alone.
Future research directions should focus on elucidating the precise molecular mechanisms connecting longevity factors to biosynthetic capacity, developing real-time monitoring systems for CLS during fermentation, and expanding these engineering strategies to additional industrial host organisms.
In the field of synthetic biology and metabolic engineering, the production of valuable compounds through heterologous expression is a cornerstone strategy. However, engineering microbial cell factories to redirect their metabolism inevitably imposes a metabolic burden on the host organism, triggering stress symptoms that include decreased growth rate, impaired protein synthesis, genetic instability, and aberrant cell size [72]. This burden stems from the fundamental reallocation of finite cellular resources—including energy, nucleotides, amino acids, and cofactors—from normal cellular processes toward the production of heterologous proteins and pathways that often provide no benefit to the host itself [73]. On an industrial scale, this cellular stress translates to processes that are not economically viable due to reduced final titers and productivity [72]. Understanding the sources of this burden and implementing strategies to manage it is therefore crucial for developing efficient biosynthetic processes. This guide objectively compares the manifestations of metabolic burden across different host systems and evaluates the experimental methodologies used to quantify and mitigate these challenges within the broader context of efficiency metrics for biosynthetic pathway research.
Comparative Analysis of Host Organisms and Their Burden Profiles
The choice of host organism fundamentally influences the nature and severity of metabolic burden. Different hosts present unique advantages and challenges concerning resource allocation, stress responses, and production capabilities.
Table 1: Comparison of Common Host Organisms for Heterologous Expression
| Host Organism | Advantages | Burden-Related Challenges | Typical Applications |
|---|---|---|---|
| Escherichia coli (Bacteria) | Fast growth, inexpensive media, high protein titers, extensive genetic tools [73] | Lack of complex post-translational modifications; strong stringent and heat shock responses to burden; inability to secrete many proteins [72] [73] | Simple therapeutic proteins, industrial enzymes [73] |
| Saccharomyces cerevisiae (Yeast) | GRAS status; eukaryotic protein processing & secretion; robust genetic tools; high-density fermentation [74] [73] [75] | Crabtree effect (ethanol production); hyperglycosylation; resource competition impacting growth and productivity [74] [73] | Biopharmaceuticals (insulin, vaccines), industrial enzymes, biofuels [74] [73] |
| Komagataella phaffii (Yeast) | GRAS status; high secretory capacity; high cell densities; Crabtree-negative; strong inducible promoters [73] [75] | Methanol use required for classic induction (toxicity, hazard); intensive engineering needed for alternative systems [73] | Pharmaceutical proteins (ecallantide), antibody fragments, industrial enzymes [73] |
| Filamentous Fungi | High native metabolite diversity; efficient protein secretion; hardy cultures [75] | Complex morphology; abundant native pathways compete for resources; potential for hazardous spores [75] | Organic acids, complex secondary metabolites, hydrolytic enzymes [75] |
Quantitative Manifestations of Metabolic Burden
The physiological impact of metabolic burden can be quantified through key performance metrics, providing researchers with data to compare hosts and engineering strategies.
Table 2: Quantitative Metrics for Assessing Metabolic Burden in Host Organisms
| Metric | Description | Measurement Techniques | Interpretation |
|---|---|---|---|
| Specific Growth Rate (μ) | The rate of biomass increase per unit time. | Optical density (OD600), dry cell weight measurements over time [72] | A decrease indicates resource diversion from growth to heterologous production. |
| Maximum Biomass Yield | The total biomass accumulated at the end of fermentation. | Final dry cell weight or final OD600 [72] | A lower yield suggests chronic burden impacting overall metabolic efficiency. |
| Heterologous Protein Titer | The concentration of the target protein produced. | HPLC, ELISA, enzymatic activity assays [74] | The primary output; high titers with minimal growth impact indicate successful burden management. |
| Product Yield on Substrate | Mass of product formed per mass of substrate consumed. | Analytics to measure substrate depletion and product formation [74] | Indicates the carbon and energy efficiency of the production process. |
| Transcriptional & Translational Load | Demand on the gene expression machinery. | RNA sequencing (RNA-seq), ribosome profiling [72] | High load can trigger stress responses and sap cellular resources. |
Experimental Protocols for Quantifying Metabolic Burden
A standardized experimental approach is vital for the objective comparison of burden across different systems. The following protocol outlines a robust methodology for assessing metabolic burden in microbial hosts.
Protocol: Growth-Based Burden Assay
Objective: To quantify the impact of heterologous pathway expression on host growth kinetics and metabolic efficiency.
Materials:
- Test Strains: Recombinant strain with heterologous pathway and an isogenic control strain (empty vector).
- Growth Medium: Defined minimal medium to precisely control nutrient availability.
- Bioreactor/Shake Flasks: For controlled aerobic cultivation.
- Analytical Instruments: Spectrophotometer (for OD600), HPLC system (for substrate and product analysis), cell counter (for viability).
Methodology:
- Inoculum Preparation: Start identical pre-cultures of both test and control strains from single colonies. Grow to mid-exponential phase.
- Main Cultivation: Dilute pre-cultures into fresh, pre-warmed medium to a standardized OD600. Perform cultivations in at least biological triplicate.
- For induced systems: Add inducer at a defined cell density.
- For constitutive systems: Monitor growth from inoculation.
- Data Collection:
- Growth Kinetics: Measure OD600 every 30-60 minutes.
- Substrate Consumption: Take samples periodically, centrifuge, and analyze supernatant via HPLC to determine glucose/glycerol depletion.
- Product Formation: Analyze supernatant or cell lysates (depending on protein localization) for target protein titer using ELISA or activity assays.
- Cell Viability: Use methylene blue staining or automated cell counters to assess culture viability at key time points.
- Data Analysis:
- Calculate the specific growth rate (μ) during the exponential phase for both strains.
- Determine the maximum biomass yield.
- Calculate the product yield on biomass (YP/X).
- Compare all parameters between the recombinant and control strains. A significant reduction in μ and/or biomass in the test strain indicates substantial metabolic burden.
This protocol provides a foundational, data-driven assessment of burden, enabling direct comparison of different engineering interventions or host systems.
Visualizing Cellular Stress Pathways Induced by Metabolic Burden
The following diagram illustrates the interconnected stress mechanisms activated in a host cell, such as E. coli, in response to the (over)expression of heterologous proteins, linking cellular triggers to their downstream symptoms.
Diagram: Cellular Stress Pathways from Heterologous Expression. This diagram maps the triggers (red), activated stress mechanisms (blue), and resulting physiological symptoms (green) that constitute "metabolic burden" in host cells like E. coli [72].
The Scientist's Toolkit: Key Reagents and Solutions
Successfully managing metabolic burden requires a suite of specialized reagents and tools. The following table details essential solutions for researching and mitigating burden in heterologous expression systems.
Table 3: Research Reagent Solutions for Metabolic Burden Management
| Reagent / Tool | Function | Application in Burden Management |
|---|---|---|
| Codon-Optimized Genes | Synthetic genes designed with host-preferred codons. | Reduces ribosomal stalling and tRNA depletion, mitigating translational burden and protein misfolding [72]. |
| Tunable Promoters | Regulatory DNA sequences with adjustable strength. | Enables fine-tuning of heterologous gene expression to find a balance between production and host fitness [74]. |
| Plasmid Vectors with Different Copy Numbers | DNA constructs that replicate at low, medium, or high copies per cell. | Allows investigation of gene dosage impact; lower copy numbers often reduce burden [72]. |
| Genome Integration Tools | Systems for inserting genes directly into the host chromosome. | Replaces high-copy plasmids, creating more genetically stable production strains with reduced replicative burden [73]. |
| Chaperone Co-expression Plasmids | Vectors expressing host-specific folding helper proteins. | Improves correct folding of heterologous proteins, reducing aggregation and the load on the native heat shock response [72]. |
| Metabolic Quiescence Inducers | Compounds that slow down core metabolism. | Shifts cellular resources from growth to production, potentially increasing yield without triggering acute stress responses [76]. |
Discussion: Integrating Metrics and Strategies for Efficient Biosynthesis
Managing metabolic burden is not a single-step intervention but a holistic exercise in systems-level engineering. The most successful strategies involve a multi-faceted approach that integrates data from the comparative and quantitative analyses described above. This includes dynamic regulation to decouple growth and production phases, systems metabolic engineering to rewire central metabolism and enhance precursor supply, and the use of consortia to distribute the burden of complex pathways across specialized strains [76] [74] [72]. The quantitative metrics provide the essential feedback for the Design-Build-Test-Learn (DBTL) cycle, allowing researchers to objectively compare the efficiency of different constructs and hosts. By viewing heterologous production through the lens of resource allocation and cellular economics, researchers can move beyond simply maximizing expression and toward optimizing the overall fitness and productivity of the cell factory, thereby achieving a sustainable balance between heterologous expression and host vitality.
Enzyme engineering represents a cornerstone of modern biocatalysis, aiming to transcend the limitations of natural enzymes for applications in therapeutics, industrial manufacturing, and sustainable technology. The dual objectives of enhancing catalytic efficiency (as measured by kcat/Km) and refining substrate specificity dominate contemporary research efforts. Where natural enzymes evolved for biological fitness, engineered enzymes must operate under industrial conditions, process non-natural substrates, and exhibit unprecedented catalytic precision. The emergence of integrated computational and experimental frameworks has revolutionized our approach to enzyme optimization, moving beyond traditional directed evolution to structure-informed and artificial intelligence (AI)-driven engineering strategies. This guide systematically compares the performance of current enzyme engineering technologies, providing experimental data and methodologies that equip researchers with practical tools for advancing biosynthetic pathway efficiency.
Performance Comparison of Engineering Platforms
The enzyme engineering landscape has diversified into complementary technological streams, each with distinct performance characteristics, limitations, and optimal application domains. The table below provides a quantitative comparison of major platforms based on benchmark results reported in recent literature.
Table 1: Performance Comparison of Modern Enzyme Engineering Platforms
| Engineering Platform | Key Performance Metrics | Typical Optimization Cycle | Key Advantages | Major Limitations |
|---|---|---|---|---|
| AI-Driven Prediction (EZSpecificity) | 91.7% accuracy identifying reactive substrates (vs. 58.3% for previous models) [77] | Computational only; experimental validation required | Exceptional prediction accuracy for substrate specificity; handles structural data | Limited to predictive tasks; requires experimental validation |
| AI-Powered Autonomous Engineering | 26-fold activity improvement (YmPhytase); 16-fold activity improvement (AtHMT) in 4 weeks [78] | 4 rounds/4 weeks | Fully autonomous DBTL cycle; minimal human intervention | Requires specialized biofoundry infrastructure; high initial setup cost |
| Deep Learning Kinetic Prediction (CataPro) | Superior accuracy and generalization for kcat, Km, and kcat/Km prediction on unbiased datasets [79] | Computational prediction only | Robust predictions across diverse enzyme families; handles mutation effects | Dependent on training data quality and coverage |
| Physics-Based Modeling & Simulation | Quantitative prediction of electrostatic effects, tunnel dynamics, and allosteric networks [80] [81] | Weeks to months for simulation and analysis | Atomistic insights; mechanism-informed designs | Computationally intensive; requires expert knowledge |
| Synzyme (Synthetic Enzyme) Engineering | Function under extreme conditions (pH, temperature) where natural enzymes fail [82] | Design, synthesis, and characterization cycles | Unprecedented environmental stability; customizable scaffolds | Limited functional complexity compared to natural enzymes |
The performance data reveals a technological maturation where computational methods significantly reduce experimental burden. AI-driven platforms demonstrate remarkable efficiency gains, with autonomous systems achieving substantial activity improvements within accelerated timeframes [78]. Prediction-focused models like EZSpecificity and CataPro provide accurate pre-screening, minimizing wasted experimental effort [77] [79]. Meanwhile, physics-based approaches offer unparalleled mechanistic insights that inform rational design strategies [80] [81].
Table 2: Comparative Performance on Specific Enzyme Engineering Challenges
| Engineering Challenge | Best-Performing Platform | Experimental Results | Key Supporting Data |
|---|---|---|---|
| Substrate Specificity Prediction | EZSpecificity (Cross-attention GNN) | 91.7% accuracy vs. 58.3% for previous model on halogenase validation [77] | Validated with 8 halogenases and 78 substrates |
| Catalytic Efficiency (kcat/Km) Enhancement | Autonomous Engineering Platform | 26-fold improvement in YmPhytase activity at neutral pH [78] | Achieved in 4 weeks with <500 variants screened |
| Kinetic Parameter Prediction | CataPro (Deep Learning) | Superior accuracy and generalization on unbiased benchmark datasets [79] | Combines ProtT5 embeddings with molecular fingerprints |
| Extreme Condition Operation | Synzyme Platforms | Retention of activity under non-physiological pH, temperature, and solvent conditions [82] | MOF-based nanozymes with peroxidase-like efficiency |
| Reaction Tunnel Optimization | MD Simulations + Rosetta | Altered substrate access and product release in cytochrome P450s [80] | Tunnel engineering guided by molecular dynamics |
Experimental Protocols for Key Methodologies
AI-Guided Enzyme Specificity Prediction
Protocol for EZSpecificity Implementation: The EZSpecificity framework employs a cross-attention-empowered SE(3)-equivariant graph neural network architecture trained on comprehensive enzyme-substrate interactions at sequence and structural levels [77]. The experimental workflow involves:
Data Preparation: Curate enzyme-substrate pairs with confirmed interaction status from structural databases (e.g., SKiD) [83]. Represent enzymes as graphs with nodes corresponding to amino acid residues and edges representing spatial proximity. Represent substrates as molecular graphs with atoms as nodes and bonds as edges.
Model Training: Implement the SE(3)-equivariant network that respects rotational and translational symmetries in 3D space [77]. The cross-attention mechanism allows the model to jointly reason about enzyme and substrate representations, capturing their mutual influence on binding and catalysis.
Validation: Employ rigorous benchmarking against held-out enzyme families not present in training data. Experimental validation should follow computational predictions using enzyme kinetics assays with purified proteins and target substrates.
Key Experimental Parameters:
- Training dataset: 13,653 unique enzyme-substrate complexes from SKiD [83]
- Validation: 8 halogenases tested against 78 substrates [77]
- Performance metric: Accuracy in identifying single potential reactive substrate
Autonomous Enzyme Engineering Workflow
The autonomous engineering platform integrates machine learning with biofoundry automation for continuous enzyme improvement [78]. The detailed protocol includes:
Initial Library Design: Generate 180 variants using a combination of protein large language model (ESM-2) and epistasis model (EVmutation) to maximize diversity and quality [78].
Automated Construction Pipeline: Implement HiFi-assembly based mutagenesis method with approximately 95% accuracy, eliminating intermediate sequencing verification steps. The workflow encompasses seven automated modules: mutagenesis PCR, DNA assembly, transformation, colony picking, plasmid purification, protein expression, and enzyme assays.
Fitness Assessment & Model Retraining: Measure variant activity using high-throughput assays appropriate for the target enzyme (e.g., methyltransferase activity for AtHMT, phosphatase activity for YmPhytase). Use resulting data to train low-N machine learning models that predict variant fitness for subsequent design cycles.
Iterative Optimization: Conduct multiple rounds of design-build-test-learn cycles (typically 3-4 rounds) with expanding library sizes based on model confidence.
Critical Implementation Details:
- Robotic integration via Thermo Momentum software
- Central robotic arm coordinates all instrumentation
- 96-well format throughout for scalability
- Fitness function must be quantifiable and automatable
Kinetic Parameter Prediction with CataPro
Implementation Protocol for Kinetic Prediction: CataPro predicts turnover number (kcat), Michaelis constant (Km), and catalytic efficiency (kcat/Km) using deep learning with enhanced generalization capability [79]. The methodology includes:
Dataset Curation: Collect enzyme kinetic entries from BRENDA and SABIO-RK databases. Apply rigorous clustering at 40% sequence similarity threshold to create unbiased benchmark datasets. Partition clusters into ten groups for cross-validation to prevent data leakage.
Feature Engineering: Encode enzyme sequences using ProtT5-XL-UniRef50 embeddings (1024 dimensions). Represent substrates using MolT5 embeddings (768 dimensions) combined with MACCS keys fingerprints (167 dimensions). Concatenate into a 1959-dimensional feature vector.
Model Architecture & Training: Implement neural network regressor with appropriate architecture for the kinetic parameter being predicted. Train on unbiased partitions with rigorous validation to prevent overfitting.
Validation Framework:
- Test on enzymes with <40% sequence similarity to training examples
- Evaluate prediction accuracy for mutant effects using deep mutational scanning data
- Experimental confirmation with newly discovered enzymes
The Scientist's Toolkit: Essential Research Reagents & Platforms
Successful implementation of modern enzyme engineering strategies requires specific computational tools, experimental platforms, and data resources. The table below catalogues essential solutions referenced in the performance data.
Table 3: Essential Research Reagent Solutions for Enzyme Engineering
| Tool/Platform | Type | Primary Function | Key Applications | Validation Evidence |
|---|---|---|---|---|
| EZSpecificity | Computational Model | Substrate specificity prediction | Identifying potential substrates for uncharacterized enzymes; predicting promiscuous activities | 91.7% accuracy in halogenase validation [77] |
| CataPro | Deep Learning Model | kcat, Km, and kcat/Km prediction | Prioritizing enzyme variants; predicting catalytic efficiency before experimental characterization | Superior performance on unbiased benchmarks [79] |
| SKiD Database | Structural Kinetics Dataset | Repository of enzyme-substrate complexes with kinetic parameters | Training predictive models; structure-function relationship studies | 13,653 unique enzyme-substrate complexes with 3D structures [83] |
| iBioFAB | Automated Biofoundry | End-to-end automated protein engineering | Executing autonomous DBTL cycles; high-throughput variant characterization | 26-fold activity improvement in YmPhytase [78] |
| Rosetta Design | Molecular Modeling Software | Enzyme active site design and optimization | Stabilizing transition states; altering substrate specificity | Successful de novo enzyme design and activity enhancement [80] |
| Molecular Dynamics | Simulation Platform | Atomic-level dynamics of enzyme conformational changes | Tunnel engineering; allosteric regulation; mechanism elucidation | Identification of key residues for catalysis and specificity [80] [81] |
The comparative performance data reveals a strategic hierarchy for enzyme engineering initiatives. AI-driven autonomous platforms deliver the most rapid efficiency gains for well-characterized enzyme systems with automatable assays [78]. For novel enzymes or those with complex kinetic characterization requirements, prediction-focused tools like EZSpecificity and CataPro provide valuable pre-screening to guide experimental efforts [77] [79]. Physics-based modeling remains essential for tackling fundamentally new catalytic challenges or when mechanistic insights are required to escape evolutionary dead ends [81].
For biosynthetic pathway optimization, the integration of multiple platforms creates a powerful engineering pipeline: prediction tools identify promising enzyme candidates, autonomous engineering enhances their catalytic properties, and synzyme approaches create custom catalysts for steps without natural enzyme solutions. This multi-layered strategy maximizes the probability of success while minimizing resource expenditure, accelerating the development of efficient biosynthetic systems for pharmaceutical, industrial, and environmental applications.
Validation Frameworks and Comparative Analysis of Pathway Performance
Sclareol, a plant-derived diterpene alcohol, serves as a valuable fragrance ingredient and a key precursor for the synthesis of ambrox, a sustainable alternative to ambergris in the perfume industry [84] [85] [86]. Traditional production methods relying on plant extraction from Salvia sclarea face significant limitations, including high costs, low yields, and environmental concerns [84] [85]. Advances in synthetic biology have enabled the development of microbial cell factories for sustainable sclareol production, with recent engineering breakthroughs achieving unprecedented titers [87] [88] [85]. This case study objectively analyzes the integrated engineering strategies that enabled sclareol production at 25.9 g/L in Saccharomyces cerevisiae, comparing this performance with alternative production platforms and providing detailed experimental protocols for replication and validation by researchers.
Performance Comparison of Sclareol Production Platforms
The quest for efficient sclareol production has employed various microbial hosts, each with distinct advantages and limitations. The table below provides a comparative analysis of reported sclareol production across different engineered systems.
Table 1: Performance Comparison of Microbial Platforms for Sclareol Production
| Host Organism | Maximum Titer (g/L) | Cultivation Scale | Key Engineering Strategies | Year | Citation |
|---|---|---|---|---|---|
| Saccharomyces cerevisiae | 25.9 | Fed-batch fermentation | Lifespan engineering + metabolic pathway optimization | 2025 | [87] [88] |
| Saccharomyces cerevisiae | 11.4 | Fed-batch fermentation | Global metabolic engineering, ERG20 modification | 2023 | [84] [85] |
| Yarrowia lipolytica | 12.9 | 5-L bioreactor | Scaffold protein for LPPS-SCS interaction optimization | 2024 | [84] [85] |
| Yarrowia lipolytica | 2.66 | Shake flask | Combinatorial metabolic engineering, lipid synthesis downregulation | 2025 | [84] [85] |
| Escherichia coli | 1.5 | Not specified | Heterologous pathway introduction | 2012 | [84] [85] |
The performance data reveals substantial progress in sclareol production capabilities, with the integrated lifespan and metabolic engineering approach in S. cerevisiae representing a remarkable improvement over previous benchmarks. This 25.9 g/L titer demonstrates a 127% increase over the previous highest reported yield in yeast and more than double the best result achieved in Y. lipolytica platforms [84] [87] [88].
Detailed Experimental Protocols
Integrated Engineering Workflow
The record-breaking sclareol production was achieved through a systematic approach combining lifespan engineering with metabolic pathway optimization. The following diagram illustrates the comprehensive experimental workflow:
Lifespan Engineering Strategies
The chronological lifespan extension involved four key dimensions of cellular engineering [87] [88]:
- Nutrient Sensing Weakening: Downregulation of conserved nutrient-sensing pathways (particularly TOR and PKA) to mimic caloric restriction effects and extend cellular lifespan.
- Mitophagy Enhancement: Engineering improved mitochondrial quality control through enhanced selective autophagy of damaged mitochondria.
- Protein Stability Optimization: Modifications to reduce accumulation of misfolded proteins and enhance proteostasis.
- Genomic Stability Enhancement: Interventions to maintain DNA integrity during prolonged fermentation.
The simultaneous implementation of nutrient sensing weakening and mitophagy enhancement proved particularly impactful, with omics analyses confirming that these interventions enhanced central metabolism and cellular robustness during later fermentation stages [87] [88].
Metabolic Pathway Engineering
The sclareol biosynthetic pathway was optimized through the following interventions:
Table 2: Metabolic Engineering Components for Enhanced Sclareol Production
| Engineering Target | Specific Interventions | Biological Impact | Experimental Evidence |
|---|---|---|---|
| MVA Pathway | Overexpression of tHMG1, ERG12, ERG8, ERG19, ERG13 | Enhanced flux to IPP/DMAPP precursors | 2.5-3.0× increase in pathway intermediates [84] |
| Acetyl-CoA Supply | Engineered TCA cycle and β-oxidation pathways | Increased central precursor availability | 40% reduction in byproduct accumulation [85] |
| GGPP Synthesis | Heterologous expression of efficient GGPPS variants | Enhanced diterpene precursor supply | 8.3× increase in GGPP levels [84] |
| Sclareol Synthase | Multi-copy integration and protein fusion strategies | Improved conversion efficiency from GGPP | 215% increase in catalytic efficiency [85] |
| Competitive Pathways | Downregulation of lipid and sterol synthesis | Redirected carbon flux toward sclareol | 60% reduction in competitive products [84] [85] |
Pathway Engineering and Metabolic Flux
The core metabolic engineering strategy focused on optimizing the mevalonate pathway while redirecting carbon flux from competitive pathways. The following diagram illustrates the key metabolic nodes targeted for engineering:
The metabolic flux was systematically redirected toward sclareol production through combinatorial engineering that enhanced precursor supply while reducing carbon diversion to native competitive pathways [84] [85].
Research Reagent Solutions
The following table details key reagents and materials essential for replicating the high-efficiency sclareol production system:
Table 3: Essential Research Reagents for Sclareol Biosynthesis Studies
| Reagent/Material | Specification/Function | Application in Sclareol Research |
|---|---|---|
| S. cerevisiae Strains | Po1f-tHEI (Y. lipolytica); BY4741 (S. cerevisiae) | Base chassis strains with enhanced MVA pathway [84] [85] |
| Plasmid Systems | pINA1312, pINA1269, CRISPRyl-Cas9 vectors | Genetic manipulation and multi-copy integration [84] [85] |
| Culture Media | YPD (Yeast Extract Peptone Dextrose); YNB (Yeast Nitrogen Base) | Strain cultivation, selection, and fermentation [84] [85] |
| Analytical Standards | Sclareol (GC ≥ 98%), GGOH (geranylgeraniol) | Quantification and method calibration [84] [85] |
| Extraction Solvent | Dodecane (10% of fermentation volume) | In-situ product extraction and volatilization reduction [84] [85] |
| Gene Modules | SsLPPS, SsSCS, tPaGGPPS (codon-optimized) | Heterologous sclareol pathway construction [84] [85] |
The integrated engineering approach combining lifespan extension with metabolic optimization represents a paradigm shift in microbial production of plant-derived terpenoids. The achievement of 25.9 g/L sclareol in S. cerevisiae demonstrates the profound impact of addressing cellular aging and metabolic stress in prolonged fermentation processes. This case study provides researchers with comprehensive experimental protocols and performance comparisons that validate the superiority of this integrated approach over conventional metabolic engineering alone. The reported strategies offer a generalizable framework for developing high-performance microbial cell factories not only for sclareol but for various high-value terpenoids and natural products, potentially transforming industrial approaches to sustainable compound production [87] [88].
The pursuit of efficient and sustainable production for high-value terpenes and phenolic acids is a central goal in modern metabolic engineering and synthetic biology. This guide objectively compares the performance of different biosynthetic strategies for these two large classes of plant specialized metabolites. The content is framed within a broader thesis on efficiency metrics for biosynthetic pathways, evaluating generalizability across products by examining pathway architecture, thermodynamic constraints, and computational prediction tools. Performance is compared through key metrics such as titer, yield, and productivity, alongside critical practical considerations like pathway complexity and scalability [89] [90] [91]. This analysis provides researchers and drug development professionals with a structured framework for selecting and optimizing production platforms.
Comparative Efficiency Analysis of Biosynthetic Pathways
The biosynthetic pathways for terpenes and phenolic acids operate on distinct biochemical logics, which directly influence their efficiency, engineering potential, and the strategies required for their multi-product validation.
Quantitative Performance Metrics
Table 1: Comparative Performance Metrics for Terpene and Phenolic Acid Biosynthesis
| Metric | Terpene Pathway (Squalene Example) | Phenolic Acid (Ferulic Acid Example) |
|---|---|---|
| Native Host Titer | 123.6 mg/L (Thraustochytrium sp.) [89] | Market size >USD 35 million; consumption >750 tons by 2025 [90] |
| Key Thermodynamic Driver | ATP investment drives pathway flux [89] | Aromatic amino acid precursor availability (L-phenylalanine) [90] [92] |
| Energy Co-factor Demand | 3 ATP, 2 NADPH per IPP (C5 unit) [89] | Not explicitly quantified, but dependent on shikimate and phenylpropanoid pathways [90] |
| Common Engineering Target | 3-Hydroxy-3-Methylglutaryl CoA Reductase (HMGR) [89] | Phenylalanine Ammonia Lyase (PAL)/Tyrosine Ammonia Lyase (TAL) [90] [93] |
| Pathway Promiscuity | High (single cyclase can produce dozens of scaffolds) [91] | Moderate (core pathway provides precursors for diverse branches) [90] [92] |
Pathway Architecture and Logic
Terpene Biosynthesis Logic: The terpene biosynthetic logic is characterized by high promiscuity and conformational flexibility. A single terpene cyclase can produce dozens of different hydrocarbon scaffolds from universal C5 precursors (IPP/DMAPP). This diversification occurs through carbocation-driven cyclizations and rearrangements held in defined conformations within the cyclase enzyme. The subsequent "tailoring" steps, often involving cytochrome P450s (CYPs) and glycosyltransferases (UGTs), further expand structural diversity, frequently exhibiting broad substrate tolerance [91]. This intrinsic promiscuity is a valuable feature for multi-product validation, as it allows for the generation of chemical diversity from a relatively simple pathway foundation.
Phenolic Acid Biosynthesis Logic: Phenolic acids are derived primarily from the shikimate and phenylpropanoid pathways, branching from the aromatic amino acids phenylalanine and tyrosine. The core pathway begins with the deamination of phenylalanine by phenylalanine ammonia-lyase (PAL) to yield trans-cinnamic acid. Further hydroxylation and methylation reactions produce a series of hydroxycinnamic acids (e.g., p-coumaric, caffeic, ferulic acids). Similarly, hydroxybenzoic acids can be derived from the shortening of the side chain of their cinnamic acid analogs [90] [92]. While the core pathway is more linear than that of terpenes, diversification occurs through specific hydroxylation, methylation, and glycosylation patterns, leading to thousands of structures, including complex flavonoids [92].
Experimental Protocols for Pathway Validation
Validating the generalizability of biosynthetic strategies requires a combination of modern omics technologies, metabolic engineering, and analytical chemistry.
Integrative Omics for Gene Discovery
For discovering novel pathway genes, especially in non-model organisms, an effective protocol involves:
- Multi-Tissue Full-Length Transcriptome Sequencing: Utilize hybrid sequencing platforms (e.g., PacBio long-read and Illumina short-read sequencing) on multiple plant organs to obtain comprehensive and accurate transcriptomic data [94].
- Phylogenetic Analysis of Enzyme Families: Identify candidate genes (e.g., Cytochrome P450s (CYPs) and UDP-glycosyltransferases (UGTs)) by constructing phylogenetic trees with reference enzymes from well-characterized species [94].
- Weighted Gene Co-Expression Network Analysis (WGCNA): Correlate gene expression patterns with the accumulation profiles of target metabolites across different tissues. Genes within the same biosynthetic pathway are often co-expressed [49] [94].
- Functional Characterization: Heterologously express and purify candidate enzymes (e.g., in E. coli) and test their activity against proposed substrates in vitro [94].
Thermodynamic Analysis of Pathway Flux
To identify and overcome thermodynamic bottlenecks in pathways like terpene biosynthesis:
- Proteomic and Metabolomic Profiling: Under different growth conditions (e.g., with/without sodium elicitation), perform comparative proteomics and measure intracellular metabolite levels to identify shifts in energy metabolism and pathway fluxes [89].
- Thermodynamic Modeling: Analyze the local thermodynamics of each pathway step. A key bottleneck in the mevalonate pathway, for instance, is the non-decarboxylative Claisen condensation of acetyl-CoA to acetoacetyl-CoA, catalyzed by acetyl-CoA acetyltransferase (ACAT) [89].
- Elicitor-Driven Flux Enhancement: Implement strategies that increase cellular ATP levels. For example, sodium elicitation in Thraustochytrium sp. was shown to increase respiration, shifting energy generation to lipid oxidation and providing the ATP needed to drive the thermodynamically constrained mevalonate pathway [89].
Biosynthetic Pathway Mapping and Experimental Workflows
The following diagrams illustrate the core biosynthetic pathways and a generalized workflow for multi-product validation.
Terpene and Phenolic Acid Biosynthesis Pathways
(Diagram 1: Core biosynthetic pathways for terpenes and phenolic acids.)
Multi-Product Validation Workflow
(Diagram 2: A generalized experimental workflow for validating biosynthetic pathways.)
The Scientist's Toolkit: Research Reagent Solutions
Successful dissection and engineering of these pathways rely on a suite of specialized reagents and computational tools.
Table 2: Essential Research Reagents and Tools for Pathway Validation
| Tool/Reagent Category | Specific Examples | Function in Validation |
|---|---|---|
| Computational Pathway Prediction | plantiSMASH [49], PhytoClust [49], Retrosynthesis Algorithms [38] | Identifies biosynthetic gene clusters (BGCs) and predicts potential biosynthetic routes from available precursors. |
| Gene Editing & Engineering | CRISPR-Cas9 [90] | Enables precise genome editing in microbial or plant hosts to knock out competing pathways or insert heterologous genes. |
| Analytical Standards | Reference phenolic acids (gallic, protocatechuic, p-coumaric, ferulic acids) [90] [93] | Essential for calibrating analytical instruments (HPLC, GC-MS) and quantifying product titer and purity. |
| Authentication & QC | DNA Barcoding [95], HPLC [95], Thin-Layer Chromatography [95] | Verifies the authenticity of plant material used for gene discovery or extraction, preventing adulteration. |
| Biosensors | Genetically Encoded Biosensors [90] | Allows high-throughput screening of microbial libraries for variants with enhanced production of target metabolites. |
Biosynthetic Gene Clusters (BGCs) are sets of co-localized genes that encode the enzymatic machinery for the production of specialized microbial metabolites, also known as natural products. These compounds, which include antibiotics, cytotoxins, siderophores, and immunosuppressants, are not essential for primary growth but provide producers with significant competitive advantages in their ecological niches, such as defense against predators, virulence, and metal acquisition [96] [97] [98]. The systematic identification and comparison of BGCs across different species—a process known as comparative genomics—enables researchers to unravel the vast, untapped potential of microbial natural products, with profound implications for drug discovery, agriculture, and understanding symbiotic relationships.
The foundation of this approach lies in the conserved biosynthetic logic of BGCs. These clusters typically contain backbone genes (e.g., for Polyketide Synthases (PKS), Non-Ribosomal Peptide Synthetases (NRPS), and terpene cyclases) that define the core structure of the metabolite, alongside tailoring enzymes, regulatory genes, and transporters that modify and export the final product [97] [98]. Comparative genomics leverages this genetic architecture by grouping BGCs into Biosynthetic Gene Cluster Families (GCFs). GCFs consist of BGCs that share significant sequence similarity and domain architecture, implying they produce structurally related chemicals [99] [96]. This methodology allows scientists to move beyond single-genome analysis to a high-resolution, cross-species perspective, revealing the evolutionary trajectories—such as gene gain, loss, and duplication—that have shaped the metabolic capabilities of organisms over millions of years [99] [97].
Computational Workflows for BGC Identification and Comparison
The initial and crucial step in comparative BGC analysis is the accurate prediction and annotation of gene clusters from genomic data. This process relies on specialized bioinformatics tools and pipelines designed to handle the complexity and diversity of biosynthetic pathways.
Table 1: Key Computational Tools for BGC Identification and Analysis
| Tool Name | Primary Function | Key Features | Applicability |
|---|---|---|---|
| antiSMASH [100] [97] | BGC Detection & Annotation | Identifies known & novel BGCs; predicts core biosynthetic enzymes & cluster classes | Bacteria & Fungi |
| plantiSMASH [49] | Plant BGC Detection | Uses plant-specific pHMMs to identify biosynthetic genes & distinguish clusters from tandem arrays | Plants |
| BiG-FAM [96] | GCF Analysis | Groups BGCs into families (GCFs) based on shared domain sequence similarity | Bacteria & Fungi |
| BioNavi-NP [43] | Retro-biosynthesis Planning | Predicts biosynthetic pathways for natural products using deep learning | Natural Product Discovery |
| SMURF [98] | Fungal BGC Prediction | Predicts fungal cluster backbone enzymes (PKS, NRPS, DMATS) | Fungi |
The standard workflow begins with the assembly of high-quality genome sequences, as assembly completeness and annotation accuracy are paramount for avoiding fragmentation of BGCs and missing genetic components [49]. For instance, in a study of Alternaria fungi, gene prediction was uniformly re-performed on all genomes using the funannotate pipeline to eliminate technical bias before BGC mining [97]. Subsequently, tools like antiSMASH are employed to scan the genomes and predict BGCs, categorizing them into major classes such as PKS, NRPS, terpene, and ribosomally synthesized and post-translationally modified peptides (RiPPs) [99] [96] [97]. The final stage involves comparative analysis, where BGCs are classified into GCFs using tools like BiG-FAM or custom similarity networks. This allows researchers to distinguish a conserved "core" set of GCFs present across all studied species from "accessory" or "singleton" GCFs that may be lineage-specific or unique to certain strains [99] [96]. Determining BGC boundaries remains a challenge, but the use of synteny—the conservation of gene order and homology across genomes—has proven an effective bioinformatics solution for predicting these borders [100].
Figure 1: A generalized computational workflow for the comparative genomic identification of Biosynthetic Gene Clusters (BGCs) and their grouping into Gene Cluster Families (GCFs).
Experimental Protocols for Validation and Characterization
In silico predictions require experimental validation to confirm the existence and function of the putative metabolites. The following protocols outline key methodologies for this crucial phase.
Protocol: Heterologous Expression and Metabolite Analysis
This protocol is used to activate silent BGCs and characterize the chemical structures of their products.
- Cluster Selection and Cloning: A prioritized BGC, often one that is ubiquitous or unique, is amplified from the genomic DNA and cloned into an expression vector suitable for a heterologous host (e.g., S. cerevisiae or Aspergillus nidulans for fungal BGCs) [96] [98].
- Transformation and Cultivation: The recombinant vector is introduced into the heterologous host. Positive transformants are selected and cultivated in an appropriate medium to induce expression [98].
- Metabolite Extraction: The culture broth and/or mycelium is extracted with organic solvents (e.g., ethyl acetate or methanol) to capture a wide range of natural products.
- Chemical Analysis and Dereplication: The crude extract is analyzed using Liquid Chromatography coupled with High-Resolution Mass Spectrometry (LC-HRMS). This step, known as dereplication, identifies known compounds and flags novel ones by matching mass spectra against natural product databases [101] [96].
- Bioactivity Testing: The crude extract or purified compounds are tested in bioassays relevant to the ecological context, such as antimicrobial activity against competing microbes or cytotoxicity against insect or mammalian cells [101] [96].
Protocol: Gene Inactivation and Phenotypic Screening
This protocol establishes a direct link between a BGC and its biological function.
- Gene Knockout: A core biosynthetic gene (e.g., a PKS or NRPS) within the target BGC is disrupted via targeted gene deletion using techniques such as CRISPR-Cas9 or homologous recombination [98].
- Metabolite Profiling: The metabolic profile of the mutant strain is compared to that of the wild-type strain using analytical techniques like Thin Layer Chromatography (TLC), High-Performance Liquid Chromatography (HPLC), or NMR. The absence of a specific compound in the mutant confirms its production by the targeted BGC [98] [96].
- Phenotypic Assay: The wild-type and mutant strains are subjected to phenotypic assays. For example, a study on Xenorhabdus and Photorhabdus bacteria tested mutants for their ability to suppress insect immune systems, directly linking a specific GCF to an ecological function [96].
Comparative Analysis of BGCs Across Kingdoms of Life
The application of these genomic and experimental strategies across different biological kingdoms reveals distinct patterns in BGC distribution, evolution, and function. The table below provides a comparative summary of key efficiency metrics derived from large-scale genomic studies.
Table 2: Efficiency Metrics in BGC Research: A Cross-Kingdom Comparison
| Study Organism | Genomes Analyzed | Total BGCs Identified | BGCs per Genome (Avg.) | Notable Findings & Conserved GCFs |
|---|---|---|---|---|
| Fungi: Termitomyces [99] | 39 | 754 | 19 - 34 | 7 core GCFs present in all 21 species; NRPS-like and terpene BGCs most abundant. |
| Fungi: Alternaria & Pleosporaceae [97] | 187 | 6,323 | 29 - 34 | Mycotoxin BGCs (e.g., alternariol) restricted to specific taxonomic sections. |
| Bacteria: Brevibacterium [101] | 98 | Not Specified | Variable | Only 2.5% core genome; BGCs (e.g., for phenazines) show clade-specific distribution. |
| Bacteria: Xenorhabdus & Photorhabdus [96] | 45 | 1,000 | 22 | NRPS BGCs are most abundant (59%); a few core GCFs produce proteasome inhibitors & virulence factors. |
| Aspergillus spp. [98] | 4 | 266 (curated) | ~66 | Manual curation of 266 clusters (PKS, NRPS, DMATS) enabled systematic discovery. |
The data reveals several key insights. First, the average number of BGCs per genome is consistently high across diverse fungal genera (~20-35), underscoring their rich biosynthetic potential. In bacteria, the number can be even higher, with Xenorhabdus and Photorhabdus averaging 22 BGCs per genome, which is two- to ten-fold higher than other related Enterobacteria [96]. Second, the concept of a "core" set of universally conserved BGCs is a recurring theme. For example, all 21 species of Termitomyces fungi shared seven core GCFs, suggesting these metabolites perform fundamental functions in the symbiosis with termites [99]. Similarly, analysis of the Brevibacterium pangenome revealed that while most BGCs are accessory, specific ones like those for siderophores and carotenoids show clade-specific distribution patterns [101]. Finally, these distribution patterns are highly informative for predicting ecological roles and guiding applications. The discovery that the alternariol mycotoxin BGC is found only in specific Alternaria sections directly informs food safety monitoring efforts [97].
Figure 2: The relationship between BGC distribution patterns (Core, Accessory, Singleton) and their inferred biological functions, as revealed by comparative genomics.
A successful comparative genomics project relies on a suite of computational and experimental reagents. The following table details key solutions and their functions.
Table 3: Research Reagent Solutions for BGC Discovery
| Reagent / Resource | Function / Application | Context of Use |
|---|---|---|
| antiSMASH Database [100] | Central repository for known & predicted BGCs; enables sequence-based searches & comparison. | Genome mining & initial BGC identification across bacteria & fungi. |
| MIBiG Repository [97] | A curated database of known BGCs with experimental evidence; used as a gold-standard reference. | Dereplication of BGCs & validation of cluster boundaries and predicted products. |
| funannotate Pipeline [97] | An integrated tool for fungal genome masking, gene prediction, and functional annotation. | Standardized genome annotation prior to BGC mining to ensure consistency and quality. |
| Heterologous Host Systems (e.g., A. nidulans) [98] | A genetically tractable host for expressing BGCs from organisms that are difficult to culture or manipulate. | Activation and characterization of silent or cryptic BGCs. |
| LC-HRMS Instrumentation [101] [96] | High-resolution analytical platform for separating and determining the precise mass of metabolites. | Chemical dereplication and structural characterization of natural products from extracts. |
Comparative genomics has fundamentally transformed the identification and study of biosynthetic gene clusters, moving the field from single-organism analysis to a comprehensive, ecosystem-level perspective. By integrating robust computational workflows with rigorous experimental validation, researchers can now efficiently map the biosynthetic landscape across entire genera, distinguishing evolutionarily conserved pathways from those that confer specific adaptive advantages. The efficiency metrics and patterns emerging from these studies—such as the identification of core GCFs in fungal symbionts and the clade-specific BGCs in bacteria—provide a powerful framework for prioritizing targets for natural product discovery. This integrated approach is accelerating the development of new therapeutic agents, informing risk assessments for mycotoxins in food supplies, and deepening our understanding of the chemical language that mediates complex biological interactions.
Flux Balance Analysis (FBA) is a cornerstone mathematical approach in systems biology used to predict the flow of metabolites through metabolic networks. By leveraging genome-scale metabolic models (GEMs) that contain all known metabolic reactions of an organism, FBA computes metabolic flux distributions at steady state, enabling researchers to predict phenotypes such as growth rates or the production of specific metabolites [102] [103]. The method's power lies in its reliance on stoichiometric constraints and mass balance, avoiding the need for difficult-to-measure kinetic parameters [102]. This constraint-based approach defines a solution space of all possible metabolic flux distributions, from which an optimal state is identified based on a specified biological objective, such as the maximization of biomass or the synthesis of a target compound [102] [103].
However, a central challenge in the field revolves around the fidelity of these computational predictions when measured against experimental yield data. While FBA has proven successful in predicting various cellular phenotypes, its accuracy is inherently tied to the appropriate selection of an objective function and the constraints applied to the model [104] [105]. This article provides a comparative analysis of FBA's predictive performance against experimental measurements, examining the methodological advances that bridge the gap between in silico forecasts and in vitro results, with a particular focus on applications in microbial metabolism and drug development.
Methodological Foundations of FBA
Core Mathematical Principles
The computational framework of FBA is built upon the stoichiometric matrix, S, an m x n matrix where m represents the number of metabolites and n the number of metabolic reactions in the network. Each element in the matrix corresponds to the stoichiometric coefficient of a metabolite in a particular reaction [102]. The fundamental equation of FBA is:
Sv = 0
This equation describes the steady-state condition, where the vector v represents the flux (reaction rate) of every reaction in the network. The system assumes that metabolite concentrations do not change over time, meaning the production and consumption of each metabolite are perfectly balanced [102] [103]. As most metabolic networks are underdetermined (more reactions than metabolites), this system has infinitely many solutions. FBA identifies a single, optimal flux distribution by imposing an objective function to be maximized or minimized, typically formulated as:
Z = cTv
Here, c is a vector of weights that defines how much each reaction contributes to the cellular objective, such as growth or product synthesis [102]. The solution is found using linear programming, subject to additional constraints that define upper and lower bounds (vmin and vmax) for each reaction flux, representing known physiological limits [102].
Workflow and Experimental Integration
The following diagram illustrates the standard FBA workflow, from model construction to the validation of predictions:
Diagram 1: The FBA Workflow. The process begins with data integration, proceeds through model setup and optimization, and concludes with experimental validation, which often informs further model refinement [102] [103].
A critical assumption in standard FBA is that the system is at steady state. While this simplifies calculations, it can limit the model's ability to capture transient metabolic dynamics. To address this, extensions like Dynamic FBA (dFBA) have been developed, which incorporate time-dependent changes in the extracellular environment [104]. Furthermore, the accuracy of predictions is highly dependent on the chosen objective function. Common choices include biomass maximization for simulating growth or the production rate of a specific metabolite for biotechnological applications [102] [103]. Selecting an objective that does not reflect the true cellular priorities is a primary source of discrepancy between predictions and experimental yields.
Advances in FBA Methodologies for Improved Predictions
Frameworks for Identifying Objective Functions
A significant frontier in FBA research is the development of methods to systematically infer objective functions from experimental data, rather than relying on a priori assumptions. The TIObjFind (Topology-Informed Objective Find) framework addresses this by integrating Metabolic Pathway Analysis (MPA) with traditional FBA [104] [106]. This approach determines Coefficients of Importance (CoIs) that quantify each metabolic reaction's contribution to an objective function, thereby aligning optimization results with experimental flux data [104]. By focusing on specific pathways rather than the entire network, TIObjFind enhances the interpretability of complex metabolic networks and provides insights into how cells adapt their metabolic objectives under different environmental conditions [104].
Another powerful machine learning-based approach is Flux Cone Learning (FCL). This method uses Monte Carlo sampling to generate a large corpus of possible flux distributions from a GEM. A supervised learning algorithm is then trained on this data, paired with experimental fitness scores from gene deletion screens, to predict the effects of genetic perturbations on cellular phenotypes [105]. A key advantage of FCL is that it "delivers best-in-class accuracy for prediction of metabolic gene essentiality" and outperforms standard FBA predictions without requiring an explicit optimality assumption, making it applicable to a wider range of organisms [105].
Integration with Machine Learning and Complex Dynamics
Machine learning is further leveraged to overcome the computational bottlenecks of integrating FBA with complex simulations, such as reactive transport models (RTMs). As demonstrated by one study, training artificial neural networks (ANNs) as surrogate FBA models can reduce computational time by several orders of magnitude while maintaining robust solutions [107]. This ANN-based approach was successfully used to simulate the metabolic switching of Shewanella oneidensis MR-1 across different carbon sources (lactate, pyruvate, acetate), a dynamic process that is challenging to capture with traditional methods [107].
For specific engineering applications, FBA models can be enhanced with enzyme constraints. The ECMpy workflow, for instance, incorporates constraints based on enzyme availability and catalytic efficiency (k~cat~ values), which prevents the model from predicting unrealistically high fluxes and improves the biological relevance of its predictions [103]. This is particularly useful in metabolic engineering, where pathway enzymes are often modified to enhance product yield.
The relationships between these advanced methodologies are summarized below:
Diagram 2: Relationships Between Advanced FBA Methodologies. Newer frameworks extend standard FBA to address specific limitations, such as unknown objectives, computational cost, and the need for kinetic data [104] [105] [107].
Comparative Analysis: FBA Predictions vs. Experimental Data
Predictive Accuracy Across Organisms and Conditions
The table below summarizes the performance of FBA and its advanced derivatives in predicting key metabolic phenotypes against experimental measurements.
Table 1: Comparison of FBA Predictions vs. Experimental Yields
| Organism / System | Prediction Method | Experimental Measurement | Key Result / Accuracy | Reference / Context |
|---|---|---|---|---|
| Escherichia coli | Standard FBA (Biomass max.) | Growth rate (Aerobic vs. Anaerobic) | Predicted growth rates: 1.65 hr⁻¹ (aerobic), 0.47 hr⁻¹ (anaerobic); agreed well with experimental data. | [102] |
| Escherichia coli | Flux Cone Learning (FCL) | Metabolic Gene Essentiality | 95% accuracy in predicting gene essentiality, outperforming standard FBA. | [105] |
| Clostridium acetobutylicum & Multi-species system | TIObjFind Framework | Pathway-specific flux data | Reduced prediction errors and improved alignment with experimental data by identifying key reaction weights (Coefficients of Importance). | [104] |
| Shewanella oneidensis MR-1 | Multi-step FBA with ANN Surrogates | Dynamic metabolic switching (Lactate→Pyruvate→Acetate) | Successfully simulated complex growth patterns and byproduct formation; required parameter optimization to match experimental byproduct levels (~70% of theoretical max). | [107] |
| Engineered E. coli (L-cysteine production) | Enzyme-Constrained FBA (ECMpy) | L-cysteine export flux | Used to predict effects of mutated enzymes (SerA, CysE) and optimize medium conditions; required lexicographic optimization to balance production with non-zero growth. | [103] |
Analysis of Discrepancies and Limitations
The comparative data reveals several common sources of divergence between FBA predictions and experimental yields:
- Incorrect Objective Functions: The assumption of a single, universal objective like biomass maximization is a major limitation. Biological objectives can shift with environmental conditions, a challenge that frameworks like TIObjFind aim to solve [104].
- Missing Network Components: Even well-curated GEMs can have gaps. For example, the iML1515 model for E. coli was found to lack key reactions for thiosulfate assimilation into L-cysteine, requiring manual "gap-filling" to enable accurate predictions [103].
- Lack of Regulatory Constraints: Standard FBA does not account for gene regulatory networks or enzyme inhibition, which can lead to overprediction of flux. Incorporating enzyme constraints, as with the ECMpy workflow, helps mitigate this issue [103].
- Computational vs. Biological Optima: A solution that is mathematically optimal for a simplified model may not be biologically feasible. Methods like FCL that learn from experimental data rather than purely optimizing a function can better capture biological reality [105].
Experimental Protocols for Validation
Protocol for Validating Gene Essentiality Predictions
This protocol is used to generate experimental data for benchmarking computational tools like FBA and FCL [105].
- Strain Construction: Create a library of single-gene knockout mutants for the organism of interest using a method like CRISPR-Cas9 or lambda Red recombination.
- Growth Assays: Culture each knockout strain and the wild-type strain in a defined medium with a specified carbon source (e.g., glucose). Use microtiter plates for high-throughput screening.
- Fitness Measurement: Monitor cell density (OD~600~) over time. The exponential growth rate (µ) of each mutant is calculated and normalized to the growth rate of the wild-type strain to determine a fitness score.
- Essentiality Classification: A gene is classified as "essential" if the fitness score of its knockout mutant is below a pre-defined threshold (e.g., unable to grow, or fitness < 0.1). Non-essential genes allow for residual growth.
- Data Comparison: The list of experimentally essential genes is compared against in silico predictions. A gene is considered correctly predicted if the model's growth rate for the corresponding knockout is zero (or near-zero) for an essential gene, and positive for a non-essential gene.
Protocol for Measuring Metabolic Fluxes for FBA Validation
Quantifying intracellular metabolic fluxes is crucial for directly validating FBA predictions beyond growth phenotypes [104] [107].
- Isotope Labeling: Grow cells in a defined medium where the sole carbon source is a ^13^C-labeled compound (e.g., ^13^C-glucose).
- Metabolite Extraction: During mid-exponential growth, rapidly harvest cells and quench metabolism. Extract intracellular metabolites.
- Mass Spectrometry Analysis: Analyze the metabolite extract using Gas Chromatography or Liquid Chromatography coupled to Mass Spectrometry (GC-/LC-MS). This measures the mass isotopomer distribution of key intermediate metabolites (e.g., amino acids, TCA cycle intermediates).
- Flux Calculation: Use computational software (e.g., INCA, ^13^C-FLUX) to fit a metabolic network model to the measured isotopomer data. This estimation procedure calculates the most probable intracellular flux map that is consistent with the experimental labeling pattern.
- Data Alignment: Compare the computed exchange fluxes and key internal fluxes (e.g., flux through pentose phosphate pathway) against the flux distribution predicted by FBA.
Table 2: Key Reagents and Computational Tools for FBA Research
| Item / Resource | Function / Application | Example / Source |
|---|---|---|
| Genome-Scale Model (GEM) | A structured database of all known metabolic reactions and genes for an organism; the core of any FBA simulation. | iML1515 (for E. coli [103]), iMR799 (for S. oneidensis [107]) |
| COBRA Toolbox | A MATLAB-based software suite for performing constraint-based reconstruction and analysis, including FBA. | [102] |
| COBRApy | A Python version of the COBRA Toolbox, enabling FBA simulations and model manipulation. | [103] |
| BRENDA Database | A comprehensive enzyme information system used to obtain kinetic parameters (e.g., k~cat~ values) for enzyme-constrained models. | [103] |
| Stoichiometric Matrix (S) | The mathematical representation of the metabolic network, defining the mass balance constraints for the model. | Constructed from GEMs [102] |
| Defined Growth Medium | A culture medium with a precise and known composition; critical for setting accurate uptake constraints in the FBA model. | M9 minimal medium with a single carbon source [102] |
| ^13^C-Labeled Substrates | Tracers used in experimental flux validation to determine intracellular reaction rates via ^13^C-Metabolic Flux Analysis (^13^C-MFA). | e.g., U-^13^C-Glucose [104] |
| Monte Carlo Sampler | An algorithm used in methods like Flux Cone Learning to randomly sample the solution space of a metabolic network for machine learning. | [105] |
The transition from laboratory-scale success to industrial-scale production represents a critical juncture in the development of biosynthetic processes. While laboratory experiments demonstrate proof-of-concept, industrial implementation demands meeting rigorous economic and operational criteria that extend far beyond simple proof-of-function. This guide objectively compares the key efficiency metrics and considerations that differentiate laboratory and industrial production environments, providing researchers with a framework for evaluating biosynthetic pathways against the demanding requirements of commercial-scale manufacturing. The analysis is situated within the broader context of optimizing efficiency metrics for biosynthetic pathways, where understanding these scale-dependent factors enables more effective research prioritization and process development.
Industrial biotechnology success hinges on developing processes that are not only scientifically sound but also economically viable and scalable. As projects advance through development stages, clinical material demand grows tremendously, and production scale-up is rarely straightforward, with time-consuming, expensive, and unexpected challenges often emerging [108]. The disconnect between microscale experimentation and macroscale production requirements can lead to promising research findings failing to translate into commercially viable processes. By understanding these scale-up considerations early in the research and development pathway, scientists can design experiments and metabolic engineering strategies that better anticipate industrial constraints, ultimately increasing the translational potential of their work.
Key Efficiency Metrics: A Comparative Analysis
The performance of biological production systems is typically evaluated through three fundamental chemical production metrics: titer, rate, and yield (TRY) [109]. However, the relative importance and acceptable thresholds for these metrics differ significantly between laboratory and industrial contexts, with additional economic and operational factors becoming critical at scale.
Table 1: Laboratory vs. Industrial Production Efficiency Metrics
| Metric | Laboratory Context | Industrial Context |
|---|---|---|
| Titer | Often reported as maximum achievable concentration (e.g., g/L) | Must be high enough to make downstream processing economical; typically >50-100 g/L for commodities |
| Productivity (Rate) | May be reported as specific productivity (g/g cells/h) | Volumetric productivity (g/L/h) is critical for facility throughput and capital cost amortization |
| Yield | Mole of product per mole of substrate; demonstrates pathway efficiency | Directly impacts raw material costs; must be economically competitive with chemical routes |
| Process Intensity | Often minimal consideration | High cell density fermentations preferred to reduce reactor volume and downstream processing costs |
| Feedstock Cost | Often uses pure substrates for consistency | Must utilize low-cost, often variable, renewable feedstocks at industrial scale |
| Byproduct Formation | Noted but not always quantified economically | Significant impact on purification costs and waste treatment expenses |
Beyond the core TRY metrics, industrial processes must contend with additional economic and operational considerations that are rarely determining factors in laboratory research. Substrate costs become critically important at scale, where the choice between pure sugars versus complex or waste-derived feedstocks can dramatically impact process economics [15]. Product purification represents a substantial portion of total production costs industrially, influenced by factors such as titer, byproduct formation, and the physical properties of the fermentation broth [110]. Utility consumption for sterilization, oxygen transfer, cooling, and agitation must be minimized through process optimization. Microbial robustness under industrial conditions—including tolerance to inhibitors, osmolality, shear stress, and product toxicity—becomes essential for consistent performance across long production campaigns [111].
Experimental Protocols for Scale-Relevant Evaluation
Advanced Adaptive Laboratory Evolution (ALE) with High-Throughput Screening
Purpose: To rapidly generate microbial strains with enhanced industrial phenotypes, particularly improved tolerance to high product concentrations and metabolic fitness under production conditions.
Methodology:
- Initial Mutagenesis: Generate a diverse genetic library of the production host (e.g., E. coli W3110) using in vivo mutagenesis (IVM) to create a pool of genetic variants, enhancing genetic diversity beyond spontaneous mutation rates [111].
- Automated Microdroplet Cultivation: Implement an automated microbial microdroplet culture (MMC) system for high-throughput cultivation within microliter-scale droplets. This system integrates serial passaging, real-time optical density monitoring, gradient-based addition of chemical stressors, and programmable droplet sorting [111].
- Biosensor-Assisted Screening: Employ product-responsive biosensors to enable high-throughput screening for individuals exhibiting both improved tolerance and maintained or enhanced production capacity, identifying advantageous "win-win" phenotypes [111].
- Validation: Characterize top-performing strains in bench-scale bioreactors to confirm performance improvements under controlled conditions.
Applications: This approach has been successfully applied to evolve E. coli strains capable of tolerating 720 mM 3-hydroxypropionic acid (3-HP) within 12 days, with the top-performing strain producing 86.3 g/L 3-HP with a yield of 0.82 mol/mol glycerol [111].
Genome-Scale Metabolic Modeling for Host Selection and Pathway Design
Purpose: To systematically evaluate and compare the metabolic capacities of different host organisms for target chemical production, enabling data-driven host selection before experimental implementation.
Methodology:
- Model Construction: Develop genome-scale metabolic models (GEMs) that represent gene-protein-reaction associations in candidate host organisms [15].
- Pathway Reconstruction: Incorporate biosynthetic pathways for target chemicals using metabolic reactions demonstrated to function properly, adding heterologous reactions when necessary [15].
- Yield Calculation: Calculate both maximum theoretical yield (YT) and maximum achievable yield (YA) for each host-pathway combination. Y_A accounts for non-growth-associated maintenance energy and minimum growth requirements, providing a more realistic assessment of metabolic capacity [15].
- Comparative Analysis: Systematically compare metabolic capacities across multiple host organisms (e.g., Bacillus subtilis, Corynebacterium glutamicum, E. coli, Pseudomonas putida, and Saccharomyces cerevisiae) under different aeration conditions and with various carbon sources [15].
- Experimental Validation: Test top-predicted host-pathway combinations in laboratory fermentations to validate model predictions.
Applications: This methodology has been applied to evaluate the metabolic capacities of five industrial microorganisms for the production of 235 different bio-based chemicals, enabling identification of the most suitable host for specific chemical production [15].
Computational Pathway Design Using SubNetX Algorithm
Purpose: To design stoichiometrically feasible, high-yield biosynthetic pathways for complex natural and non-natural compounds by extracting and ranking balanced metabolic subnetworks.
Methodology:
- Reaction Network Preparation: Define a database of elementally balanced reactions, target compounds, and precursor compounds, incorporating both known biochemical reactions and computationally predicted reactions [19].
- Graph Search: Identify linear core pathways from precursor compounds to target compounds using graph-search algorithms [19].
- Subnetwork Expansion: Expand and extract a balanced subnetwork where cosubstrates and byproducts are linked to the native metabolism, ensuring stoichiometric feasibility [19].
- Host Integration: Integrate the subnetwork into the genome-scale metabolic model of the host organism to verify production capability within host metabolic constraints [19].
- Pathway Ranking: Rank feasible pathways based on yield, enzyme specificity, thermodynamic feasibility, and the number of heterologous steps required [19].
Applications: The SubNetX algorithm has been successfully applied to design pathways for 70 industrially relevant natural and synthetic chemicals, including complex pharmaceuticals, demonstrating the ability to identify pathways with higher production yields compared to linear pathways [19].
Visualizing the Scale-Up Workflow
The transition from laboratory discovery to industrial production follows a structured pathway with distinct evaluation criteria at each stage. The following diagram illustrates this workflow, highlighting key decision points and optimization targets.
Diagram 1: Biosynthetic pathway scale-up workflow from laboratory validation to industrial production, highlighting key optimization stages and the evolution of critical evaluation metrics.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful scale-up of biosynthetic processes requires specialized reagents, tools, and methodologies at each development stage. The following table details key solutions that facilitate the transition from laboratory research to industrial implementation.
Table 2: Key Research Reagent Solutions for Biosynthetic Pathway Development and Scale-Up
| Reagent/Tool | Function | Scale Relevance |
|---|---|---|
| Automated Microdroplet Culture (MMC) Systems | Enable high-throughput cultivation and evolution with real-time monitoring and programmable sorting | Laboratory |
| Product-Responsive Biosensors | Allow high-throughput screening for strains balancing improved tolerance with production capacity | Laboratory |
| Genome-Scale Metabolic Models (GEMs) | Computational platforms for predicting metabolic capacity, host suitability, and engineering targets | Laboratory to Pilot |
| Platform Strains | Pre-engineered microbial chassis with optimized central metabolism for chemical production | Laboratory to Industrial |
| Cell-Free DNA Synthesis Systems | Enable rapid production of DNA constructs without bacterial sequences or antibiotic resistance genes | Laboratory to Pilot |
| Combinatorial Biosynthesis Tools | Methods for combining natural and engineered enzymes from disparate sources into modified pathways | Laboratory to Industrial |
| Specialized Excipients (e.g., HPC, HPMC, PVP) | Improve flowability and processability of high drug-load formulations during manufacturing | Pilot to Industrial |
| Advanced Tooling Coatings (CrN, TiN) | Reduce sticking and adherence during tablet compression, improving manufacturing efficiency | Industrial |
Platform strains represent a particularly valuable tool in scale-up considerations. These are pre-engineered microbial chassis with optimized central metabolism, developed to allow easy insertion of different product formation pathways, thereby significantly reducing development time [109]. For example, engineered versions of E. coli and S. cerevisiae have been developed as platform strains for the production of diverse chemical targets, providing a foundation of favorable traits that can be further customized for specific applications [109] [15].
Cell-free synthesis systems are gaining importance for both research and production applications. These systems use cell lysates prepared in bulk that can be thawed and used immediately, eliminating the need for strain propagation and simplifying downstream processing since unnecessary cellular components are not included [112]. The ability to rapidly implement cell-free protein synthesis reactions is also driving its use in the rapid evaluation of novel biosynthetic pathways and genetic circuits before commitment to full cellular engineering [112].
The successful translation of biosynthetic pathways from laboratory demonstration to industrial production requires careful attention to the evolving priorities and constraints across development stages. While laboratory research rightly focuses on demonstrating scientific feasibility through metrics like titer, yield, and productivity, industrial implementation introduces additional dimensions including economic viability, operational robustness, and scalability. By incorporating scale-up considerations early in the research and development process—through strategic host selection, computational modeling, and directed evolution of industrially relevant phenotypes—researchers can significantly enhance the translational potential of their work. The tools and methodologies outlined in this guide provide a framework for bridging the gap between laboratory innovation and industrial implementation, ultimately supporting the development of more efficient and economically viable biomanufacturing processes.
Conclusion
The optimization of biosynthetic pathway efficiency requires an integrated approach combining foundational metrics with advanced computational and biological strategies. Recent breakthroughs in lifespan engineering, demonstrated by 25.9 g/L sclareol production, and AI-driven pathway design tools like BioNavi-NP, have significantly accelerated the development of high-performance microbial cell factories. The successful application of these strategies across diverse product classes confirms their generalizability for pharmaceutical and industrial applications. Future directions should focus on dynamic pathway regulation, machine learning-guided enzyme engineering, and host chassis development to further enhance metabolic flux and cellular robustness. These advances promise to transform biosynthetic production into a more predictable, efficient, and economically viable platform for next-generation drug development and sustainable chemical manufacturing.
References
- 1. BioTRY: A Comprehensive Knowledge Base for Titer, Rate, ... [https://pubmed.ncbi.nlm.nih.gov/39423319/]
- 2. Genome-scale metabolic rewiring improves titers rates and ... [https://www.nature.com/articles/s41467-020-19171-4]
- 3. an efficient strain design strategy for balanced yield, titer, and ... [https://bmcbiotechnol.biomedcentral.com/articles/10.1186/1472-6750-13-8]
- 4. Metabolic engineering of Escherichia coli for high-yield ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12175537/]
- 5. Engineering Non-Natural Biosynthetic Pathways for ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717625001697]
- 6. Step-by-step optimization of a heterologous pathway for de ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11316701/]
- 7. MCF2Chem: A manually curated knowledge base of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10625697/]
- 8. Stress-Induced Biological Aging: A Review and Guide for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10243290/]
- 9. Cellular Models of Aging and Senescence [https://www.mdpi.com/2073-4409/14/16/1278]
- 10. Metabolic modelling reveals the aging-associated decline ... [https://www.nature.com/articles/s41564-025-01959-z]
- 11. Metabolic Biomarkers for Lifespan Extension and Cellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12567969/]
- 12. Comparative machine learning strategies for improving ... [https://www.sciencedirect.com/science/article/pii/S0168160525003885]
- 13. The science of fermentation (2025) | GFI [https://gfi.org/science/the-science-of-fermentation/]
- 14. Compatibility engineering of synthetic metabolic pathways ... [https://www.sciencedirect.com/science/article/abs/pii/S0009250925014447]
- 15. Comprehensive evaluation of the capacities of microbial ... [https://www.nature.com/articles/s41467-025-58227-1]
- 16. Enhanced microbial production of pyridoxine (Vitamin B6) ... [https://pubmed.ncbi.nlm.nih.gov/41089825/]
- 17. Pathway optimization and membrane engineering for ... [https://www.sciencedirect.com/science/article/abs/pii/S0960852425013823]
- 18. Development of an efficient heterologous protein expression ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02786-x]
- 19. Designing pathways for bioproducing complex chemicals ... [https://www.nature.com/articles/s41467-025-59827-7]
- 20. Hierarchical metabolic engineering for rewiring cellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12514951/]
- 21. In silico profiling of Escherichia coli and Saccharomyces ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/1475-2859-12-84]
- 22. Saccharomyces cerevisiae vs Escherichia coli in the ... [https://www.sciencedirect.com/science/article/abs/pii/S2589014X23003055]
- 23. Enhancing Biofuel Production Through Escherichia coli ... [https://www.mdpi.com/2227-9717/13/7/2115]
- 24. An outlook to sophisticated technologies and novel ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2023.1249841/full]
- 25. Engineering Saccharomyces cerevisiae for medical applications [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02625-5]
- 26. Metabolic engineering of Escherichia coli for squalene ... [https://www.sciencedirect.com/science/article/pii/S2405805X25000882]
- 27. Metabolic Engineering of Saccharomyces cerevisiae - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC98985/]
- 28. The Growing Scope of Applications of Genome-scale ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3108568/]
- 29. Review Genetic circuits for metabolic flux optimization [https://www.sciencedirect.com/science/article/pii/S0966842X24000040]
- 30. Advances in the optimization of central carbon metabolism in metabolic engineering | Microbial Cell Factories | Full Text [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-023-02090-6]
- 31. Evolution-guided optimization of biosynthetic pathways - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4273373/]
- 32. Establishing a versatile toolkit of flux enhanced strains and cell extracts for pathway prototyping - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1096717623001489]
- 33. Engineering a suite of E. coli strains for enhanced expression of bacterial polysaccharides and glycoconjugate vaccines | Microbial Cell Factories | Full Text [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-022-01792-7]
- 34. Enhanced flux potential analysis links changes in enzyme ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11965317/]
- 35. A computational workflow for the expansion of ... [https://www.nature.com/articles/s41467-021-22022-5]
- 36. Computer-aided design and implementation of efficient ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2024.1360740/full]
- 37. brsynth/rp2paths: RetroPath2.0 to pathways [https://github.com/brsynth/rp2paths]
- 38. computational methods for biosynthetic pathway design [https://www.sciencedirect.com/science/article/pii/S2405805X25000687]
- 39. novoStoic2.0: An integrated framework for pathway synthesis ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1012516]
- 40. Computer-aided design and implementation of efficient ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11228882/]
- 41. Computational scoring and experimental evaluation of ... [https://www.nature.com/articles/s41587-024-02214-2]
- 42. Structural basis of potent inhibition of the human CAD ... [https://www.sciencedirect.com/science/article/abs/pii/S0006291X25015207]
- 43. Deep learning driven biosynthetic pathways navigation for ... [https://www.nature.com/articles/s41467-022-30970-9]
- 44. Graph-sequence enhanced transformer for template-free ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12365517/]
- 45. Review of Machine Learning Methods for the Prediction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8247443/]
- 46. computational methods for biosynthetic pathway design [https://pmc.ncbi.nlm.nih.gov/articles/PMC12159830/]
- 47. From data to discovery: leveraging big data in plant natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12183336/]
- 48. Multi-omics Data Integration, Interpretation, and Its Application [https://pmc.ncbi.nlm.nih.gov/articles/PMC7003173/]
- 49. Integrative omics approaches for biosynthetic pathway ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9491492/]
- 50. Multi-omic network inference from time-series data [https://www.nature.com/articles/s41540-025-00591-1]
- 51. Network-based multi-omics integrative analysis methods in ... [https://biodatamining.biomedcentral.com/articles/10.1186/s13040-025-00442-z]
- 52. Genomic prediction powered by multi-omics data [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1636438/full]
- 53. The State Of: Multi-Omics [https://frontlinegenomics.com/the-state-of-multi-omics/]
- 54. Metabolomics and transcriptomics based multi-omics ... [https://www.nature.com/articles/s41540-023-00305-5]
- 55. Single-cell differential expression analysis between ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12343076/]
- 56. Differential gene expression analysis pipelines and ... [https://www.sciencedirect.com/science/article/pii/S2001037024000424]
- 57. Comprehensive Analysis of Tissue-wide Gene Expression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5928498/]
- 58. Comparative multi-tissue profiling reveals extensive ... [https://elifesciences.org/articles/78556]
- 59. an interactive tool for pathway guided visualization of ... [https://pubmed.ncbi.nlm.nih.gov/40581591/]
- 60. Visualization methods for differential expression analysis [https://www.rna-seqblog.com/visualization-methods-for-differential-expression-analysis/]
- 61. Engineering chronological lifespan toward a robust yeast ... [https://pubmed.ncbi.nlm.nih.gov/41213019/]
- 62. Researchers Boost Biosynthetic Capacity in Yeast Through ... [https://english.cas.cn/newsroom/research_news/chem/202511/t20251117_1115869.shtml]
- 63. Regulation of Autophagy and Mitophagy by Nutrient ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3969632/]
- 64. The mitophagy pathway and its implications in human ... [https://www.nature.com/articles/s41392-023-01503-7]
- 65. Evolution-guided optimization of biosynthetic pathways [https://pubmed.ncbi.nlm.nih.gov/25453111/]
- 66. Suppressing false positive enrichment during biosensor- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9375551/]
- 67. Evolution-guided Bayesian optimization for constrained ... [https://www.nature.com/articles/s41524-024-01274-x]
- 68. Synthetic biology and metabolic engineering paving the way ... [https://pubs.rsc.org/en/content/articlehtml/2025/ya/d5ya00118h]
- 69. Balancing Cell Growth and Product Synthesis for Efficient ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12561436/]
- 70. Yeast cells experience chronological life span extension ... [https://www.sciencedirect.com/science/article/pii/S2405844025012794]
- 71. Extension of chronological life span in yeast by decreased ... [https://genesdev.cshlp.org/content/20/2/174.abstract]
- 72. stress symptoms and its related responses induced by (over ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02370-9]
- 73. Burden Imposed by Heterologous Protein Production in ... [https://www.frontiersin.org/journals/fungal-biology/articles/10.3389/ffunb.2022.827704/full]
- 74. Engineering strategies for enhanced heterologous protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10801990/]
- 75. Heterologous Metabolic Pathways: Strategies for Optimal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7385092/]
- 76. Relieving metabolic burden to improve robustness and ... [https://www.sciencedirect.com/science/article/pii/S0734975024000958]
- 77. Enzyme specificity prediction using cross-attention graph neural networks | Nature [https://www.nature.com/articles/s41586-025-09697-2]
- 78. A generalized platform for artificial intelligence-powered ... [https://www.nature.com/articles/s41467-025-61209-y]
- 79. Robust enzyme discovery and engineering with deep learning using CataPro | Nature Communications [https://www.nature.com/articles/s41467-025-58038-4]
- 80. Cutting-edge computational approaches in enzyme design and activity enhancement - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1369703X24002973]
- 81. Physics-based Modeling in the New Era of Enzyme Engineering [https://pmc.ncbi.nlm.nih.gov/articles/PMC12239909/]
- 82. Synzymes: The Future of Modern Enzyme Engineering [https://link.springer.com/article/10.1007/s12010-025-05305-1]
- 83. A structure-oriented kinetics dataset of enzyme-substrate interactions | Scientific Data [https://www.nature.com/articles/s41597-025-05829-5]
- 84. Combinatorial metabolic engineering of Yarrowia lipolytica for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12082891/]
- 85. Combinatorial metabolic engineering of Yarrowia lipolytica for ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02744-7]
- 86. Sclareol and linalyl acetate are produced by glandular ... [https://www.nature.com/articles/s41438-021-00640-w]
- 87. Researchers boost biosynthetic capacity in yeast through ... [https://scientificinquirer.com/2025/11/19/researchers-boost-biosynthetic-capacity-in-yeast-through-extended-lifespan/]
- 88. A combination of lifespan and metabolic engineering boosts ... [https://window-to-china.de/2025/11/18/a-combination-of-lifespan-and-metabolic-engineering-boosts-sclareol-yields-in-yeast/]
- 89. ATP Drives Efficient Terpene Biosynthesis in Marine ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8262955/]
- 90. Advances and Prospects of Phenolic Acids Production ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7356249/]
- 91. BJOC - Bacterial terpene biosynthesis: challenges and ... [https://www.beilstein-journals.org/bjoc/articles/15/283]
- 92. Phenolics Biosynthesis - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/phenolics-biosynthesis]
- 93. Photosynthetic apparatus efficiency, phenolic acid profiling ... [https://www.nature.com/articles/s41598-021-83695-y]
- 94. Effective prediction of biosynthetic pathway genes involved ... [https://www.nature.com/articles/s42003-022-03000-z]
- 95. Advancing herbal medicine: enhancing product quality and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10561302/]
- 96. Global analysis of biosynthetic gene clusters reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9177418/]
- 97. Genome mining reveals the distribution of biosynthetic gene ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11754-z]
- 98. Comprehensive annotation of secondary metabolite ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/1471-2180-13-91]
- 99. Comparative genomics unravels a rich set of biosynthetic ... [https://www.nature.com/articles/s42003-024-06887-y]
- 100. Determining biosynthetic gene cluster boundaries through ... [https://pubmed.ncbi.nlm.nih.gov/40651826/]
- 101. Exploring the biosynthetic gene clusters in Brevibacterium [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09694-7]
- 102. What is flux balance analysis? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3108565/]
- 103. Flux Balance Analysis | Virginia - iGEM 2025 [https://2025.igem.wiki/virginia/fba]
- 104. Optimization-based framework with flux balance analysis (FBA ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013635]
- 105. Accurate prediction of gene deletion phenotypes with Flux ... [https://www.nature.com/articles/s41467-025-63436-9]
- 106. Optimization-based framework with flux balance analysis ... [https://pubmed.ncbi.nlm.nih.gov/41144574/]
- 107. Coupling flux balance analysis with reactive transport ... [https://www.nature.com/articles/s41598-025-89997-9]
- 108. SCALE-UP & MANUFACTURING - Smart Formulation ... [https://drug-dev.com/scale-up-manufacturing-smart-formulation-processing-engineering-solutions-to-solve-drug-product-scale-up-manufacturing-challenges-with-minimum-to-no-regulatory-impact/]
- 109. Impact of synthetic biology and metabolic engineering on ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975015000361]
- 110. Engineering microbial cell factories for the production of plant ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-017-0732-7]
- 111. A Refined Adaptive Laboratory Evolution Strategy With ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12591173/]
- 112. Biosynthetic Methods in Pharmaceutical Manufacturing [https://www.biopharminternational.com/view/biosynthetic-methods-in-pharmaceutical-manufacturing]