From Primary Metabolism to Powerful Medicines: Harnessing Biosynthetic Building Blocks for Drug Discovery
This article explores the critical role of biosynthetic building blocks derived from primary metabolism in the creation of bioactive natural products.
From Primary Metabolism to Powerful Medicines: Harnessing Biosynthetic Building Blocks for Drug Discovery
Abstract
This article explores the critical role of biosynthetic building blocks derived from primary metabolism in the creation of bioactive natural products. Tailored for researchers, scientists, and drug development professionals, it provides a comprehensive analysis of the foundational principles, methodological applications, and current challenges in the field. We examine how primary metabolites like amino acids, acyl-CoAs, and nucleotides serve as precursors for complex secondary metabolites with therapeutic potential. The content covers advanced strategies in synthetic biology and combinatorial biosynthesis for optimizing production, discusses analytical and computational tools for pathway validation, and synthesizes key takeaways to outline future directions for biomedical and clinical research, offering a holistic perspective on this essential interface of metabolism and medicine.
The Chemical Blueprint of Life: Exploring Primary Metabolites as Biosynthetic Precursors
The traditional dichotomy between primary and secondary metabolism is a concept rooted in the historical development of biochemistry. Albrecht Kössel's 1891 definition separated the universal, "necessary for life" primary metabolites from the "random or not necessary" secondary metabolites [1]. However, contemporary research reveals this distinction to be increasingly artificial, demonstrating instead a deeply integrated metabolic continuum where primary metabolism supplies the essential building blocks for the vast chemical diversity of secondary metabolites [1] [2]. This in-depth technical guide explores the fundamental linkages between these metabolic domains, framing them within advanced biosynthetic building blocks research critical for drug discovery and development. For researchers and scientists, understanding this interface is paramount for harnessing the biosynthetic potential of living organisms, particularly for the sustainable production of valuable natural products with pharmacological activity [3] [4].
The following core diagram illustrates the foundational relationship between primary metabolic pathways and the major classes of secondary metabolites they support.
Diagram 1: Biosynthetic Link Between Primary and Secondary Metabolism.
Defining the Metabolic Domains
Comparative Analysis of Metabolic Types
The following table summarizes the defining characteristics of primary and secondary metabolites, highlighting their distinct yet interconnected roles.
Table 1: Characteristic Differences Between Primary and Secondary Metabolites [5] [6]
| Basis for Comparison | Primary Metabolites | Secondary Metabolites |
|---|---|---|
| Definition & Role | Directly involved in growth, development, and reproduction; essential for survival [6]. | Not directly involved in primary processes; essential for ecological interactions (defense, competition) [5] [7]. |
| Universal Presence | Found in all living organisms without exception [1] [5]. | Distribution is often species-specific or restricted to certain phylogenetic groups [5]. |
| Production Phase | Produced during the growth phase (trophophase) [6]. | Typically produced during the stationary phase (idiophase) or in response to stress [7] [6]. |
| Chemical Diversity | Limited diversity; includes universal macromolecules (proteins, nucleic acids, carbohydrates, lipids) [6]. | Extremely high chemical diversity; includes alkaloids, terpenoids, phenolics, and glucosinolates [1] [7]. |
| Quantity Produced | Produced in large quantities [6]. | Produced in small quantities [6]. |
| Function in Research | Used in various industries (food, biofuels) [6]. | Valued for pharmacological activities; used in drug development and agrochemicals [3] [5]. |
The Building Block Paradigm: From Primary Precursors to Secondary Products
Primary metabolism generates a pool of core metabolites that serve as universal biosynthetic building blocks. These precursors are funneled into specialized secondary metabolic pathways, often via gatekeeping enzymes that mark the transition point between the two metabolic domains [8]. The major building blocks and their secondary product families are summarized below.
Table 2: Primary Metabolite Building Blocks and Their Secondary Product Families [5] [7] [2]
| Primary Metabolite Building Block | Biosynthetic Origin | Major Classes of Secondary Metabolites | Key Examples (Pharmacological Use) |
|---|---|---|---|
| Acetyl-CoA / Intermediates from Glycolysis & MEP Pathway | Krebs Cycle, Glycolysis, Plastidial MEP Pathway [1] [7] | Terpenoids (Monoterpenes, Diterpenes, Triterpenes) | Paclitaxel (anticancer) [5], Artemisinin (antimalarial) [7], Gibberellins (plant hormone) [1] |
| Amino Acids (e.g., Tryptophan, Tyrosine, Lysine, Aspartate) | Nitrogen Assimilation & Primary Metabolism [5] | Alkaloids (various sub-classes) | Morphine (analgesic) [5], Vincristine (anticancer) [1] [5], Nicotine (insecticide) [5] |
| Phenylalanine / Shikimate Pathway Intermediates | Shikimate Pathway [5] | Phenolic Compounds (Flavonoids, Lignin, Tannins) | Flavonoids (antioxidants) [7] [6], Salicylic Acid (anti-inflammatory) [5], Lignin (structural polymer) [5] |
| β-Nicotinamide Adenine Dinucleotide (β-NAD) | Nucleotide Metabolism [8] | Novel β-NAD-derived Natural Products | Altemicidin, SB-203208 (isoleucyl-tRNA synthetase inhibitor) [8] |
Advanced Experimental Protocols for Elucidating Metabolic Links
Protocol 1: Gene Cluster Activation and Metabolite Identification in Bacteria
This methodology details the process of identifying novel secondary metabolites derived from primary metabolic building blocks, as demonstrated in the discovery of β-NAD-derived natural products [8].
Objective: To elucidate the biosynthetic pathway of unusual secondary metabolites with unknown primary metabolite precursors.
Workflow Overview:
Diagram 2: Experimental Workflow for Novel Pathway Elucidation.
Detailed Methodology:
- Resistance Gene-Guided Genome Mining: Identify a Biosynthetic Gene Cluster (BGC) of interest through genome sequencing and bioinformatics analysis, focusing on clusters with low homology to known systems [8].
- Heterologous Expression: Clone the entire BGC into a suitable microbial host, such as Streptomyces lividans TK21, to activate the expression of the pathway [8].
- Gatekeeping Enzyme Identification via Single Gene Expression: Construct single gene expression strains for each uncharacterized gene in the cluster. Subject the culture extracts to untargeted metabolomics analysis (e.g., LC-MS) under various analytical conditions to identify accumulating intermediates [8].
- Intermediate Structure Elucidation: Isolate the accumulating metabolite of high polarity. Use NMR spectroscopy and high-resolution mass spectrometry to determine its chemical structure, which may reveal an unexpected primary metabolite origin [8].
- Substrate Screening & In Vitro Reconstitution: Based on the intermediate's structure, hypothesize potential primary metabolite substrates (e.g., β-NAD, S-adenosylmethionine). Incubate recombinant gatekeeping enzyme with candidate substrates and co-factors to validate the initial enzymatic transformation [8].
- Downstream Pathway Reconstitution: Express and purify the remaining biosynthetic enzymes. Reconstitute the entire pathway in vitro to confirm the sequence of reactions leading to the final natural product and characterize the function of each novel enzyme [8].
Protocol 2: Multi-Omics Interrogation of Plant Metabolic Reprogramming
This protocol is used to decode the interplay between primary and secondary metabolism in plants in response to abiotic stress or elicitors [9] [7].
Objective: To understand the metabolic reprogramming and transcriptional regulation that links primary metabolic flux to the biosynthesis of specialized secondary metabolites under stress conditions.
Detailed Methodology:
- Controlled Stress/Elicitor Application: Subject model plants (e.g., alfalfa, white clover) or non-model medicinal species (e.g., Epimedium pubescens, Polygonatum kingianum) to defined abiotic stress (e.g., UV-B, drought, salinity) or treat with signaling molecules (e.g., melatonin, diethyl aminoethyl hexanoate - DA-6) or nanoparticles (Selenium NPs) [1] [9].
- Integrated Sample Collection: Harvest plant tissues at multiple time points post-treatment for parallel transcriptomic, metabolomic, and physiological analyses.
- Transcriptome Sequencing & Analysis: Perform RNA-seq on test and control samples. Identify Differentially Expressed Genes (DEGs), with a focus on transcription factors (e.g., WRKY) and genes encoding core biosynthetic enzymes for both primary and secondary metabolism [9] [7].
- Non-Targeted Metabolome Profiling: Analyze the same tissue extracts using GC-MS and LC-MS platforms. Identify and quantify Differentially Accumulated Metabolites (DAMs), covering both primary (sugars, amino acids, organic acids) and secondary (alkaloids, terpenoids, phenolics) metabolites [9].
- Multi-Omics Data Integration: Perform correlation network analysis to link gene expression patterns with metabolite accumulation. Construct co-expression networks to identify key regulatory nodes and potential metabolons (enzyme complexes) [4]. This step is crucial for mapping primary metabolic shifts to the induction of specific secondary metabolic pathways.
- Functional Validation: Clone and characterize candidate genes (e.g., OsDUF868.12 for salt tolerance) via overexpression and/or knockdown studies. Validate enzyme function in vitro and confirm metabolite production in engineered systems [9].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Technologies for Metabolic Link Research
| Research Reagent / Technology | Function & Application in Metabolic Research |
|---|---|
| Stable Isotope-Labeled Precursors (e.g., ¹³C-Glycerol, ¹âµN-Aspartate, Dâ‚‚O) | Used in isotopic labelling experiments to trace the incorporation of primary metabolites into secondary metabolic scaffolds, establishing definitive biosynthetic routes [8]. |
| Heterologous Expression Systems (e.g., Streptomyces lividans, S. cerevisiae, Plant Hairy Root Cultures) | Serve as programmable biofactories to express silent or complex BGCs, produce problematic intermediates, and elucidate pathways without interference from the native host's metabolism [8] [2]. |
| Biosynthetic Gene Clusters (BGCs) Predictors (e.g., antiSMASH, PRISM) | Bioinformatics tools for the in silico identification of genomic loci encoding secondary metabolic pathways, providing the first step in linking genes to chemistry [3]. |
| Gatekeeping Enzymes (e.g., Terpene Synthases, SbzP-like Aminotransferases, Polyketide Synthases) | Key enzymatic targets for research as they catalyze the first committed step from primary metabolic pools into secondary pathways; their study reveals novel biochemical transformations [8]. |
| Signaling Molecule Elicitors (e.g., Melatonin, Methyl Jasmonate, Hydrogen Sulfide, Nitric Oxide) | Used to mimic stress conditions and activate the endogenous regulatory networks that control the flux from primary to secondary metabolism, boosting the production of target compounds [1] [7]. |
| Metabolon Engineering Tools (CRISPR-Cas, Synthetic Scaffolds) | Emerging approaches to spatially organize sequential enzymes of a pathway to enhance channeling of primary precursors into desired secondary products, minimizing off-target effects and increasing yield [4]. |
| 1-(2,3-Dichlorphenyl)piperazine | 1-(2,3-Dichlorphenyl)piperazine, CAS:41202-77-1, MF:C10H12Cl2N2, MW:231.12 g/mol |
| Gly6 | Gly6, CAS:3887-13-6, MF:C12H20N6O7, MW:360.32 g/mol |
The historical view of secondary metabolism as a dispensable adjunct to primary metabolism has been conclusively overturned. Modern research underscores a deeply integrated system where primary metabolites serve as essential building blocks for a vast arsenal of specialized compounds critical for an organism's survival and ecological interaction [1] [7]. For drug development professionals, the implications are profound: understanding the genetic and enzymatic links that govern the flow of carbon and nitrogen from primary to secondary metabolism provides unprecedented control over the biosynthetic machinery.
Future research will be dominated by efforts to decode the spatial organization of metabolism within cells, understanding how metabolons (transient enzyme complexes) enhance pathway efficiency [1] [4]. The integration of artificial intelligence and deep learning will accelerate the prediction of BGC functions and the design of optimized enzymes [3] [4]. Furthermore, the discovery of entirely new classes of building blocks, such as β-NAD [8], suggests that our current knowledge of the metabolic inventory is still incomplete. Continued exploration of this interface, powered by the advanced experimental and computational tools outlined in this guide, promises to unlock a new generation of natural product-based therapeutics and sustainable bioprocesses.
Within every living cell, a concise set of primary metabolites serves as the universal chemical feedstock for life's diverse molecular structures. For researchers and drug development professionals, a deep understanding of these core building blocks—their biosynthetic origins and the tools to study them—is fundamental to advancing metabolic engineering and natural product discovery. This guide provides a technical examination of the essential primary metabolic pathways, detailing the key metabolites they produce, the experimental methodologies used to elucidate their flow, and the computational frameworks employed to navigate biosynthetic networks. Framed within the context of contemporary biosynthetic building block research, this resource serves as a toolkit for manipulating metabolic pathways to innovate therapeutic development [10] [11].
The Core Primary Metabolic Pathways and Their Products
Primary metabolism converts simple precursors into the essential building blocks for cellular machinery. The following table summarizes the major pathways and their key metabolite outputs.
Table 1: Essential Primary Metabolic Pathways and Key Building Blocks
| Metabolic Pathway | Key Precursor Metabolites | Primary Building Blocks Produced | Derived Product Classes |
|---|---|---|---|
| Glycolysis & Gluconeogenesis | Glucose, Phosphoenolpyruvate (PEP) [10] | Pyruvate, Glycerol-1-phosphate [11], 3-Phosphoglycerate [11] | Sugars, Polysaccharides, Glycerol backbone of lipids [11] |
| Shikimate Pathway | Phosphoenolpyruvate (PEP), Erythrose 4-Phosphate (E4P) [10] | Chorismate [10], Shikimate [10] | Aromatic Amino Acids (Phenylalanine, Tyrosine, Tryptophan) [10], Plant-derived antibiotics & pigments [10] |
| Tri-carboxylic Acid (TCA) Cycle | Acetyl-CoA | α-Ketoglutarate, Succinyl-CoA, Oxaloacetate | Amino Acids (Glutamate family), Heme, Tetrapyrroles |
| Mevalonate (MVA) / Methylerythritol Phosphate (MEP) Pathways | Acetyl-CoA [12] | Isopentenyl pyrophosphate (IPP), Dimethylallyl pyrophosphate (DMAPP) [12] | Terpenoids, Steroids [13] |
| Amino Acid Biosynthesis | Various intermediates from Glycolysis, TCA, Shikimate | 20 Proteinogenic Amino Acids [13] [10] | Non-ribosomal peptides (NRPs), Alkaloids [13] |
The following diagram illustrates the interconnectedness of these primary metabolic pathways and the key building blocks they generate.
Experimental Methodologies for Pathway Elucidation
Elucidating the flow of metabolites from primary building blocks to complex products requires a suite of sophisticated experimental protocols.
Genome Mining and Heterologous Expression
The identification of Biosynthetic Gene Clusters (BGCs) is the foundational first step. antiSMASH (antibiotics and Secondary Metabolite Analysis Shell) is the predominant tool for BGC identification in genomic data [14] [15]. Following identification, heterologous expression is used to validate BGC function. The protocol involves cloning the entire BGC into a model host organism, such as Streptomyces lividans, which does not produce the compound natively. Successful production of the target metabolite in the heterologous host confirms the identity and functionality of the BGC, as demonstrated in the elucidation of the moenomycin A pathway [16].
Metabolite Profiling and Isotopic Labeling
Metabolite profiling using Mass Spectrometry (MS) provides a snapshot of the metabolic state of a system. Coupling MS with separation techniques like chromatography (LC-MS/MS) allows researchers to separate, detect, and quantify thousands of metabolites in a single run [15]. For tracing the incorporation of primary metabolites into complex pathways, isotopic labeling is indispensable. The methodology involves feeding cells with a (^{13}\text{C})- or (^{14}\text{C})-labeled precursor (e.g., (^{13}\text{C})-glucose). The fate of the labeled atom is then tracked using NMR or MS, allowing for the precise mapping of biosynthetic pathways, as historically used to determine the non-mevalonate origin of the isoprenoid chain in moenomycin [16].
Integrated Paired Omics Analysis
A powerful contemporary approach is the systematic integration of genomic and metabolomic data. The Paired Omics Data Platform (PoDP) is a community resource that facilitates the linking of public metabolomics datasets to their genomic origins [15]. The workflow involves:
- Data Generation: Sequencing the genome and acquiring MS/MS metabolomic data from the same biological sample.
- Data Deposition: Submitting genomic data to repositories like GenBank and metabolomic data to platforms like GNPS-MassIVE.
- Data Linking: Registering the paired datasets in the PoDP with minimal metadata, creating standardized genome-metabolome links.
- Correlation Analysis: Using computational tools to correlate the presence or expression of specific BGCs (the genotype) with the detection of specific molecular families in MS data (the chemotype), a process known as metabologenomics [14] [15].
Computational and Bioinformatics Toolkits
The complexity of metabolic networks necessitates advanced computational tools for prediction and analysis.
Retrobiosynthesis Prediction with Deep Learning
BioNavi-NP is a deep learning-driven tool that predicts biosynthetic pathways for natural products in a retrosynthetic manner. It uses transformer neural networks trained on general organic and biosynthetic reactions to predict plausible precursor molecules for a target compound. Through an AND-OR tree-based planning algorithm, it then iterates this process to map multi-step routes back to fundamental building blocks from the AA/MA, MVA/MEP, CA/SA, and AAs pathways [13]. This tool represents a significant advance over conventional rule-based approaches, demonstrating a 1.7-fold higher accuracy in recovering reported building blocks [13].
Large-Scale Genomic Analysis
To explore biosynthetic diversity across thousands of genomes or metagenomes, tools like BiG-SCAPE (Biosynthetic Gene Similarity Clustering And Prospecting Engine) are essential. BiG-SCAPE performs large-scale sequence similarity network analysis of BGCs, grouping them into Gene Cluster Families (GCFs) based on a combined metric of Pfam domain content, synteny, and sequence identity [14]. This allows researchers to prioritize BGCs for discovery based on their novelty or distribution. For deeper phylogenetic analysis, CORASON (CORe Analysis of Syntenic Orthologues to prioritize Natural product gene clusters) can be used to elucidate evolutionary relationships within and across GCFs [14].
Table 2: Essential Research Reagent Solutions for Metabolic Pathway Analysis
| Research Reagent / Tool | Function / Application | Key Characteristics |
|---|---|---|
| antiSMASH [14] [15] | Identification & annotation of Biosynthetic Gene Clusters (BGCs) in genomic data. | Rule-based, supports a wide range of BGC classes (PKS, NRPS, RiPPs, etc.). |
| BioNavi-NP [13] | De novo prediction of biosynthetic pathways for natural products. | Deep learning (transformer) model; 1.7x more accurate than rule-based methods. |
| BiG-SCAPE & CORASON [14] | Large-scale comparative analysis & phylogenomics of BGCs. | Groups BGCs into families (GCFs); elucidates evolutionary relationships. |
| Paired Omics Data Platform (PoDP) [15] | Community resource for linking genomic and metabolomic datasets. | Facilitates metabologenomics; enables FAIR (Findable, Accessible, Interoperable, Reusable) data principles. |
| Uniformly (^{13}\text{C})-labeled Internal Standards [17] | Normalization and quantitative analysis in spatial metabolomics. | Cost-effective; addresses physico-chemical complexity of metabolite detection. |
| Heterologous Host Systems (e.g., S. lividans) [16] | Expression of BGCs in a tractable, surrogate organism. | Confirms BGC function; enables production of novel derivatives. |
| Z-D-Chg-OH | Z-D-Chg-OH, CAS:69901-85-5, MF:C16H21NO4, MW:291.34 g/mol | Chemical Reagent |
| H-Lys(Z)-OMe.HCl | H-Lys(Z)-OMe.HCl, CAS:27894-50-4, MF:C15H23ClN2O4, MW:330.81 g/mol | Chemical Reagent |
The synergistic use of these experimental and computational tools creates a powerful workflow for discovering and engineering metabolic pathways, as visualized below.
Plant secondary metabolism represents a sophisticated biochemical landscape where simple building blocks from primary metabolism are transformed into a vast array of specialized compounds. Terpenoids, alkaloids, and phenolics constitute three major architectural classes of these specialized metabolites, each with distinct biosynthetic origins and structural frameworks [18]. These compounds play crucial ecological roles in plant defense, communication, and adaptation while offering immense therapeutic potential for drug development [19] [18]. Understanding their biosynthetic blueprints—how fundamental carbon skeletons are assembled from primary metabolic precursors—provides the foundational knowledge necessary for manipulating their production through metabolic engineering and synthetic biology approaches [20] [21]. This review systematically examines the architectural principles governing the formation of these valuable compounds, focusing on their metabolic origins, structural diversification, and experimental characterization methodologies relevant to pharmaceutical research and development.
Biosynthetic Building Blocks and Pathways
Metabolic Origins and Carbon Skeletons
The architectural diversity of plant secondary metabolites arises from the strategic diversion of primary metabolic intermediates into specialized biosynthetic pathways. Table 1 summarizes the core building blocks and basic carbon skeletons that define each major class of specialized metabolites.
Table 1: Architectural Foundations of Major Plant Specialized Metabolite Classes
| Metabolite Class | Primary Metabolic Building Blocks | Basic Carbon Skeleton | Representative Structures |
|---|---|---|---|
| Terpenoids [19] [21] | Acetyl-CoA (MVA pathway); Pyruvate & G3P (MEP pathway) | C5 (Isoprene unit); C10, C15, C20, C30, C40 chains | Monoterpenes (e.g., limonene), Sesquiterpenes (e.g., artemisinin), Diterpenes (e.g., paclitaxel) |
| Phenolics [19] [18] | Phosphoenolpyruvate & Erythrose-4-phosphate | C6-C3 (Phenylpropanoid); C6-C1 (Benzoic acid); C6-C2-C6 (Flavonoid) | Simple phenolics, Flavonoids, Lignans, Tannins |
| Alkaloids [18] | Various amino acids (e.g., tyrosine, tryptophan, lysine, ornithine) | Heterocyclic structures containing nitrogen | Indole alkaloids (e.g., mitragynine), Isoquinoline alkaloids (e.g., morphine) |
The biosynthetic grid of plant specialized metabolism originates from three central metabolic hubs: the mevalonate (MVA) and methylerythritol phosphate (MEP) pathways for terpenoids; the shikimic acid pathway for phenolics; and various amino acid metabolic pathways for alkaloids [19] [18]. The MVA pathway, conserved in eukaryotes and some archaea, utilizes acetyl-CoA to produce the universal five-carbon terpenoid precursors isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) [20] [21]. Concurrently, the MEP pathway, predominant in prokaryotes and plant plastids, generates IPP and DMAPP from pyruvate and glyceraldehyde-3-phosphate (G3P) [21]. For phenolic compounds, the shikimate pathway bridges carbon metabolism from phosphoenolpyruvate (glycolysis) and erythrose-4-phosphate (pentose phosphate pathway) to aromatic amino acids, which subsequently serve as precursors for diverse phenolic skeletons [18]. Alkaloid biosynthesis draws primarily on nitrogen-containing amino acid precursors such as tyrosine, tryptophan, lysine, and ornithine, which undergo decarboxylation and complex rearrangement to form characteristic heterocyclic structures [18].
Pathway Visualization and Metabolic Cross-Talk
The following diagram illustrates the interconnected biosynthetic routes from primary metabolic precursors to the architectural cores of terpenoids, phenolics, and alkaloids.
This metabolic map reveals the strategic diversion of primary metabolic intermediates into the specialized metabolic pathways. The MVA and MEP pathways converge on the synthesis of IPP and DMAPP, the universal C5 building blocks for terpenoid diversity [20] [21]. The shikimate pathway provides the phenylpropanoid backbone (C6-C3) that serves as the foundation for phenolic compound structural elaboration [19] [18]. Meanwhile, multiple branches of amino acid metabolism give rise to nitrogen-containing heterocyclic scaffolds characteristic of alkaloids [18]. This metabolic architecture enables plants to generate immense chemical diversity from a limited set of primary metabolic precursors.
Experimental Methodologies for Biosynthetic Pathway Characterization
Transcriptome Mining and Heterologous Expression
Elucidating complete biosynthetic pathways for plant secondary metabolites requires integrated experimental approaches. Transcriptome mining has emerged as a powerful initial step for identifying candidate biosynthetic genes in non-model plants with rich specialized metabolomes [22]. The standard workflow begins with RNA extraction from metabolically active tissues, followed by high-throughput sequencing using both short-read (Illumina) and long-read (Oxford Nanopore) technologies to ensure comprehensive transcript coverage [22]. The resulting sequences are assembled into a reference transcriptome and annotated using tools like InterProScan and BLAST against curated databases (e.g., SwissProt) to identify genes encoding key biosynthetic enzymes based on conserved domains and functional annotations [22].
Following gene identification, heterologous expression in tractable host systems enables functional characterization of putative biosynthetic enzymes. Common expression platforms include Escherichia coli for prokaryotic enzymes and Saccharomyces cerevisiae (yeast) or Nicotiana benthamiana for eukaryotic enzymes requiring post-translational modifications or subcellular compartmentalization [20] [22]. For functional screening, candidate genes are typically cloned into appropriate expression vectors and introduced into the host system, often with rate-limiting enzymes from precursor pathways (e.g., HMGR from the MVA pathway or DXS from the MEP pathway) to enhance precursor availability and product detection [22]. Metabolite production is then analyzed using gas chromatography-mass spectrometry (GC-MS) or liquid chromatography-mass spectrometry (LC-MS) by comparing retention times and mass spectra with authentic standards [23] [22].
Metabolic Engineering and Pathway Optimization
Once key biosynthetic enzymes are characterized, metabolic engineering approaches enable pathway optimization for enhanced metabolite production. Strategic interventions include modulating the expression of rate-limiting enzymes through strong promoters, engineering feedback-insensitive enzyme variants to circumvent endogenous regulation, and implementing dynamic control systems to balance metabolic flux [20] [24]. In microbial systems, this often involves the overexpression of terpene synthases coupled with enhancement of the MVA or MEP pathways to increase precursor supply [20]. In plant systems, Agrobacterium-mediated transformation has been successfully employed to engineer terpenoid biosynthesis in tobacco hairy roots, demonstrating the metabolic plasticity of plant systems for producing diverse glycosylated terpenoid derivatives [20] [24].
Recent advances have incorporated computational and artificial intelligence technologies for the rational design of high-performance cell factories, enabling predictive optimization of enzyme combinations and cultivation parameters for maximizing terpenoid yields [20]. Additionally, directed evolution approaches applied to terpene synthases have successfully overcome catalytic efficiency limitations, as demonstrated by a 30% increase in artemisinin biosynthesis through optimization of sesquiterpene cyclase activity [24].
Research Reagent Solutions for Biosynthetic Studies
Table 2: Essential Research Reagents for Secondary Metabolite Biosynthesis Studies
| Reagent/Category | Specific Examples | Research Application | Key Functions |
|---|---|---|---|
| Cloning & Expression Vectors | pHREAC plant expression vector [22] | Heterologous expression in Nicotiana benthamiana | Gateway-compatible vector for rapid cloning of biosynthetic genes |
| Enzyme Substrates | Farnesyl pyrophosphate (FPP) [22]; Geranyl pyrophosphate (GPP) [21] | In vitro enzyme assays | Core substrates for sesquiterpene and monoterpene synthases, respectively |
| Analytical Standards | Limonene, α-pinene, caryophyllene, R-linalool [22]; Mitragynine, 7-hydroxymitragynine [23] | Metabolite identification and quantification | GC-MS and LC-MS standards for compound identification and quantification |
| Critical Enzymes | HMGR (3-hydroxy-3-methylglutaryl-CoA reductase) [22]; DXS (1-deoxy-D-xylulose-5-phosphate synthase) [20] | Metabolic pathway engineering | Rate-limiting enzymes in MVA and MEP pathways, respectively; enhance precursor flux |
| Chromatography Materials | C18 columns [25]; UHPLC systems [25]; GC-MS systems [22] | Metabolite separation and analysis | High-resolution separation and detection of specialized metabolites |
Structural Diversification and Functional Consequences
Enzymatic Modifications and Decorative Reactions
The fundamental carbon skeletons described in Section 2 undergo extensive structural elaboration through various enzyme-catalyzed modifications that significantly expand their chemical diversity and functional properties. For terpenoids, the basic scaffolds produced by terpene synthases (TPS) are further modified by cytochrome P450 oxygenases (CYP450s) that introduce oxygen functional groups through hydroxylation, epoxidation, and other oxidative transformations [21]. These modifications dramatically alter the biological activity, solubility, and volatility of the parent terpenoid scaffolds.
Phenolic compounds experience perhaps the most diverse array of decorative modifications, including glycosylation (addition of sugar moieties), acylation (addition of acyl groups), prenylation (addition of prenyl chains), and methylation [19]. These modifications influence the reactivity, bioavailability, and subcellular localization of phenolic compounds. For example, glycosylation of flavonoids enhances their water solubility and storage in vacuoles, while acylation can alter their antioxidant properties and interaction with cellular membranes [19].
Alkaloids similarly undergo extensive functionalization through oxidation, reduction, methylation, and glycosylation reactions that modulate their biological activity and physicochemical properties [23] [18]. The dose-dependent bioactivity of alkaloids makes these structural modifications particularly significant for their pharmacological applications, where subtle changes to molecular structure can dramatically alter potency and selectivity [18].
Structure-Function Relationships in Bioactivity
The structural diversity within each metabolite class directly influences their biological functions and therapeutic potential. For phenolic compounds, antioxidant activity is strongly influenced by molecular structure, particularly the number and position of hydroxyl groups on the aromatic rings and the presence of extended conjugation systems that stabilize the resulting phenoxyl radicals [19] [18]. The redox chemistry of phenolics enables them to function as both antioxidants and pro-oxidants depending on concentration and cellular context, contributing to their roles in stress protection and therapeutic applications [19].
In terpenoids, structural features such as carbon skeleton type, stereochemistry, and functional groups determine their biological activities and ecological functions [19] [21]. Monoterpenes with volatile properties serve as ecological signals in plant-insect interactions, while more complex diterpenes and triterpenes with higher molecular weights and increased functionalization often exhibit potent pharmacological activities, as demonstrated by the anticancer drug paclitaxel (diterpene) and the immunomodulator ginsenoside (triterpene) [20] [18].
For alkaloids, the presence of basic nitrogen atoms incorporated into heterocyclic ring systems enables interactions with neurotransmitter receptors and ion channels, underlying their diverse pharmacological effects on the nervous system [18]. The spatial arrangement of functional groups around these nitrogen-containing scaffolds creates complementary surfaces for binding to biological targets, explaining why subtle stereochemical differences can dramatically alter potency and selectivity [23] [18].
The architectural classes of terpenoids, alkaloids, and phenolics represent nature's sophisticated solution to generating chemical diversity from a limited set of primary metabolic building blocks. The systematic diversion of acetyl-CoA, amino acids, and sugar phosphates into specialized metabolic pathways creates distinct carbon skeletons that are further elaborated through enzyme-catalyzed modifications to produce immense structural variety [19]. This chemical diversity directly enables the multifunctional bioactivities that make these compounds invaluable as pharmaceutical agents, nutraceuticals, and fragrance compounds [18].
Understanding these architectural principles provides the foundation for rational manipulation of secondary metabolic pathways through metabolic engineering and synthetic biology approaches [20] [24]. Current challenges in the field include overcoming metabolic bottlenecks in heterologous production systems, understanding the regulatory networks that control pathway flux in native producers, and elucidating the structure-activity relationships that connect molecular architecture to biological function [20] [24]. Future research directions will likely focus on integrating multi-omics data with machine learning approaches to predict pathway regulation and enzyme function, enabling more precise engineering of production platforms for high-value natural products [20]. Additionally, exploring the ecological and evolutionary drivers of structural diversity will continue to provide insights into the selective pressures that shape these complex metabolic networks in plants [19]. As these architectural principles become increasingly well-understood, they will accelerate the development of sustainable production systems for plant-derived pharmaceuticals and other valuable specialized metabolites.
Plants have long served as a cornerstone of both traditional and modern medicine, representing one of the major reservoirs of medicinal compounds [4]. The evolution of natural product discovery spans from ancient practices of using plant extracts to the contemporary era of pathway elucidation, where researchers decode the complex biosynthetic routes nature uses to assemble these valuable compounds. This journey reflects a fundamental shift from simply isolating compounds to comprehensively understanding and engineering their production systems.
This evolution is particularly significant when framed within the context of biosynthetic building blocks from primary metabolism. Primary metabolites—including amino acids, sugars, vitamins, and organic acids—are essential for growth, development, and reproduction, acting as the foundational carbon and nitrogen sources for cellular processes [26]. In contrast, secondary metabolites (also called specialized or natural products) are not directly involved in essential physiological processes but play crucial ecological roles and often exhibit remarkable pharmacological activities [8] [27].
The connection between these metabolic realms is fundamental: specialized metabolites are metabolically derived from the primary metabolite pool and assembled by distinct enzyme families [8]. Typical natural product classes like terpenoids, polyketides, or non-ribosomal peptides are derived from oligoprenyl diphosphates, activated C2-building blocks like malonyl-CoA, or amino acids [8]. Understanding how nature converts these primary metabolite building blocks into complex chemical frameworks through dedicated biosynthetic machinery represents the modern frontier of natural product discovery.
The Historical Foundation: Plant Extracts and Early Isolation Techniques
Traditional approaches to natural product discovery relied heavily on the bioactivity-guided fractionation of plant extracts. Early natural products research focused on isolating active compounds from medicinal plants used in traditional healing systems worldwide. This process typically involved harvesting plant material, creating crude extracts using various solvents, and then using pharmacological screening to identify bioactive fractions for further purification.
These classical biochemical methods included activity assays of crude protein extracts, isotope labeling of metabolites, synthetic oligodeoxynucleotide hybridization probes, homology-based cloning, and expressed sequence tags library sequencing [28]. For instance, radioisotope-labeled feeding approaches were successfully employed in elucidating pathways like galanthamine biosynthesis [28]. While these methodologies provided the foundation for our understanding of plant natural products, they were often labor-intensive and provided limited insight into the complete biosynthetic pathways or the genetic basis of production.
The major limitation of these early approaches was their inability to efficiently connect the chemical structures of natural products with the genetic information responsible for their biosynthesis. Each discovered compound represented a piece of the puzzle, but the complete picture of how plants transformed simple primary metabolites into complex molecular architectures remained largely elusive.
The Modern Revolution: Multi-Omics and Pathway Elucidation
The emergence of next-generation sequencing (NGS) in the late 2000s revolutionized the natural products landscape, providing comprehensive omics datasets that transformed pathway discovery from a piecemeal process to a systems-level science [28]. This shift enabled researchers to move beyond simply identifying what compounds plants produce to understanding how they produce them at a genetic, enzymatic, and regulatory level.
Core Omics Technologies in Pathway Elucidation
Modern pathway elucidation leverages multiple high-throughput technologies that generate vast datasets for comprehensive analysis [28] [29]. The table below summarizes the key omics technologies and their specific applications in natural product discovery.
Table 1: Multi-Omics Technologies in Natural Product Pathway Discovery
| Technology | Data Output | Application in Pathway Discovery | Representative Elucidated Pathways |
|---|---|---|---|
| Genomics | DNA sequences, gene content, chromosomal organization | Gene cluster identification, synteny analysis, phylogenetic distribution of pathways | Vinblastine, colchicine, strychnine [28] |
| Transcriptomics | Gene expression levels, co-expression networks | Identification of coordinately regulated genes, correlation with metabolite abundance | Etoposide, colchicine, strychnine, triterpene [28] |
| Metabolomics | Metabolite profiles, chemical structures, abundances | Correlation of metabolite accumulation with gene expression, identification of pathway intermediates | Galanthamine, monoterpene indole alkaloids [28] |
| Single-Cell Omics | Cell-type specific expression and metabolite data | Resolution of spatial organization of pathways within tissues | Various pathways at cell-type resolution [28] |
Computational Tools for Data Integration
The enormous volume and intricacy of genomics, transcriptomics, and metabolomics data require robust tools for data management and mining [28]. These computational approaches have become indispensable for extracting meaningful insights from large, complex, and high-dimensional datasets.
Table 2: Computational Approaches for Biosynthetic Pathway Elucidation
| Analytical Approach | Specific Tools/Methods | Function in Pathway Discovery |
|---|---|---|
| Co-expression Analysis | Pearson correlation, self-organizing maps | Identifies genes with coordinated expression across conditions |
| Homology-Based Discovery | OrthoFinder, KIPEs, BLAST search | Finds evolutionarily related genes with known functions |
| Gene Cluster Identification | ClusterFinder, antiSMASH | Identifies physically grouped genes in genomes |
| Machine Learning | Various supervised ML algorithms | Predicts gene functions and pathway components from patterns |
Experimental Workflows: From Gene Discovery to Pathway Validation
The elucidation of complete biosynthetic pathways requires the integration of multiple experimental strategies in a systematic workflow. The following diagram illustrates the comprehensive multi-omics approach that has become standard in the field.
Figure 1. Integrated Multi-Omics Workflow for Pathway Elucidation. This flowchart illustrates the comprehensive approach from sample collection to complete pathway reconstitution, highlighting the integration of multiple data types and validation strategies.
Detailed Methodologies for Key Experiments
Heterologous Expression Systems
Heterologous expression involves introducing candidate biosynthetic genes into surrogate host organisms to test their function. The most common systems include:
- Escherichia coli bacteria: Ideal for expressing prokaryotic genes and performing enzymatic assays with purified proteins [28].
- Saccharomyces cerevisiae yeast: Suitable for eukaryotic genes requiring post-translational modifications and for pathway reconstruction [28].
- Nicotiana benthamiana tobacco: Used for Agrobacterium-mediated transient expression, allowing rapid co-expression of multiple plant genes without stable transformation [28].
The Agrobacterium-mediated transient expression in N. benthamiana has particularly accelerated functional characterization of plant biosynthetic enzymes. Compared to E. coli or yeast, this approach allows for rapid and simultaneous co-expression of multiple metabolic genes with significantly less effort in engineering and optimizing the cloning platform [28].
Isotopic Labeling Experiments
Feeding experiments with isotope-labeled precursors (e.g., ¹³C, ²H, ¹âµN) remain crucial for tracing the incorporation of primary metabolites into secondary metabolite scaffolds. The protocol involves:
- Preparing labeled precursors (e.g., L-aspartic acid, glycerol) in appropriate solvents
- Feeding to plant cell cultures or enzyme assays at relevant developmental stages
- Extracting metabolites after specific time intervals
- Analyzing incorporation patterns using LC-MS or NMR techniques
- Mapping labeled atoms to specific positions in the final natural product structure
In the discovery of ß-NAD-derived natural products, isotopic labeling experiments revealed significant label incorporation for L-aspartic acid and glycerol, providing crucial clues about the primary metabolic origins of the 6-azatetrahydroindane scaffold [8].
Gene Silencing Approaches
For confirming gene function in planta, several silencing approaches are employed:
- Virus-Induced Gene Silencing (VIGS): Using modified viruses to deliver gene fragments that trigger RNA silencing of endogenous genes
- RNA Interference (RNAi): Stable transformation with constructs producing dsRNA targeting specific genes
- CRISPR-Cas9: Creating knockout mutations in target genes to observe metabolic consequences
These approaches allow researchers to connect gene function with metabolite production in the native plant context, providing essential validation of proposed biosynthetic roles.
Case Studies: Successful Pathway Elucidation
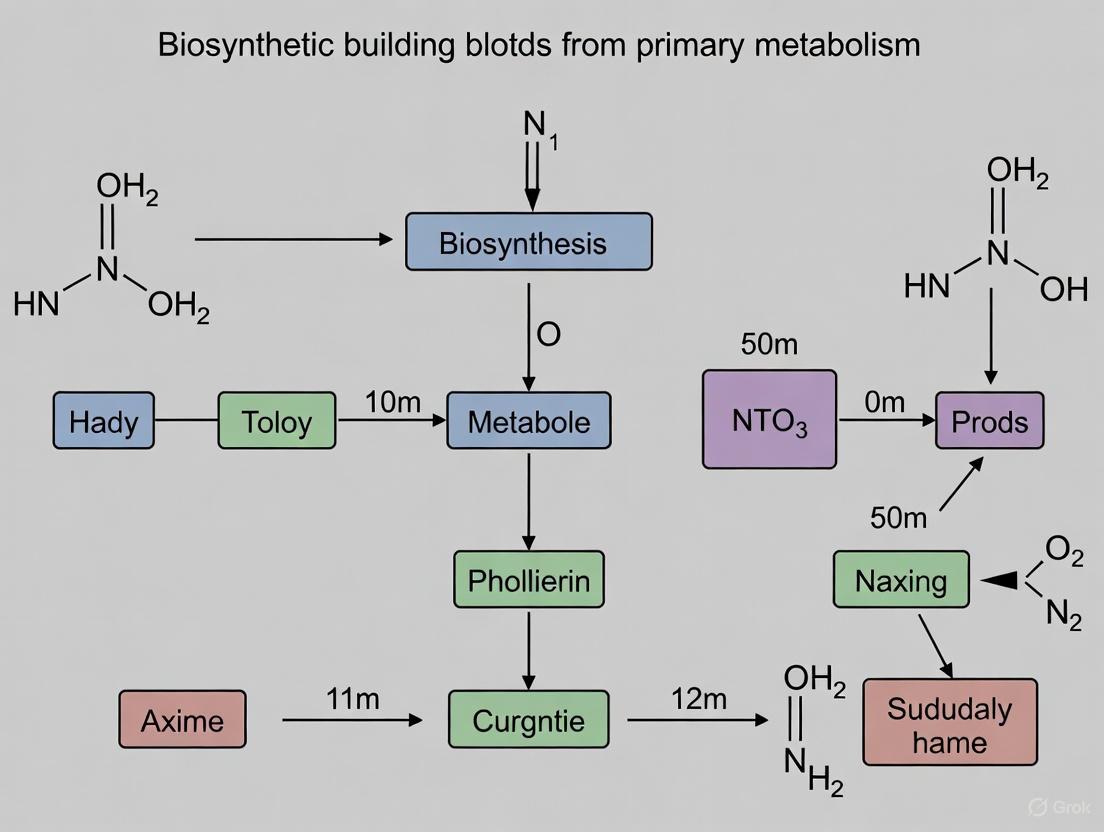
The Discovery of ß-NAD as a Natural Product Building Block
A groundbreaking discovery in the field revealed that the pivotal primary metabolite ß-nicotinamide adenine dinucleotide (ß-NAD) can function as a building block for natural product biosynthesis, establishing a novel link between primary and secondary metabolism [8]. This case study exemplifies how innovative approaches can uncover entirely new biochemical paradigms.
Researchers investigating the biosynthesis of altemicidin, SB-203207, and SB-203208—compounds with a unique 6-azatetrahydroindane scaffold—employed a combination of genomic mining, heterologous expression, and untargeted metabolomics. The key breakthrough came when they constructed single gene expression strains in the heterologous host Streptomyces lividans TK21 and subjected culture extracts to metabolomic analysis, leading to identification of a highly polar metabolite that revealed an unexpected nucleotide metabolic origin [8].
The gatekeeping enzyme SbzP was found to catalyze an unprecedented PLP-mediated tandem Cα/Cγ-alkylation reaction, leading to cyclopentane annulation at the pyridinium moiety of ß-NAD through a (3+2)-cycloaddition reaction. This represents the first enzyme known to specifically tailor ß-NAD for natural product biosynthesis [8]. The following diagram illustrates this novel biochemical transformation.
Figure 2. Novel ß-NAD-Dependent Biosynthetic Pathway. This simplified pathway shows the unprecedented use of the primary metabolite ß-NAD as a building block for natural product biosynthesis.
Complete Pathway Elucidations of Plant Natural Products
Several complex plant natural product pathways have been completely elucidated through integrated omics approaches:
Vinblastine and vincristine: These anticancer monoterpene indole alkaloids from Catharanthus roseus involve approximately 30 enzymatic steps. Their elucidation combined genomic, transcriptomic, and metabolomic data, with co-expression analysis playing a crucial role in identifying missing pathway components [28].
Strychnine: The biosynthetic pathway of this complex alkaloid from Strychnos nux-vomica was reconstructed using chemical logic-informed prediction combined with omics data. Researchers used previously elucidated steps of geissochizine oxidation as starting points, predicting that the pathway includes decarboxylation, oxidation, and reduction steps [28].
Colchicine: The complete biosynthetic pathway for this antimitotic agent was assembled using co-expression analysis of transcriptomic data from Gloriosa superba, combined with heterologous reconstitution in Nicotiana benthamiana [28].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Modern natural product pathway discovery relies on a sophisticated array of research tools and reagents. The following table details key solutions essential for conducting this research.
Table 3: Essential Research Reagents and Solutions for Natural Product Pathway Discovery
| Reagent/Solution Category | Specific Examples | Function and Application |
|---|---|---|
| Sequencing Kits | DNA library prep kits, RNA-seq kits | Generation of genomic and transcriptomic libraries for high-throughput sequencing |
| Metabolomics Standards | Stable isotope-labeled internal standards, reference compounds | Quantification and identification of metabolites in complex mixtures |
| Cloning Systems | Gateway technology, Golden Gate assembly, T4 DNA ligase | Construction of expression vectors for candidate genes |
| Heterologous Host Systems | E. coli strains, S. cerevisiae strains, N. benthamiana plants | Functional expression and characterization of biosynthetic enzymes |
| Protein Purification Kits | Affinity chromatography resins, His-tag purification systems | Isolation of recombinant enzymes for biochemical characterization |
| Enzyme Assay Reagents | Cofactors (NADPH, PLP, SAM), substrate analogs | In vitro functional characterization of enzyme activities |
| Gene Silencing Reagents | VIGS vectors, RNAi constructs, CRISPR-Cas9 components | Functional validation of genes in planta through silencing |
| Analytical Standards | Authentic natural product standards, labeled precursors | Identification and quantification of pathway intermediates |
| N-(4-Carboxycyclohexylmethyl)maleimide | trans-4-(Maleimidomethyl)cyclohexanecarboxylic Acid | High-purity trans-4-(Maleimidomethyl)cyclohexanecarboxylic acid for research use only (RUO). A key intermediate for cross-linking reagents like SMCC. Not for human or veterinary use. |
| Methioninol | L-Methioninol|CAS 2899-37-8|Research Chemical | L-Methioninol (C5H13NOS), 99+% purity. A key chiral building block for organic synthesis and biochemical research. For Research Use Only. Not for human or veterinary use. |
Future Directions and Emerging Technologies
The field of natural product discovery continues to evolve rapidly, with several emerging technologies poised to further transform our approach to pathway elucidation.
Artificial Intelligence and Machine Learning
AI and ML are playing increasingly crucial roles in predicting gene functions, pathway components, and metabolic networks from complex omics datasets [4] [28]. Supervised machine learning approaches have already been successfully applied to pathway discovery for tropane alkaloids, monoterpene indole alkaloids, and benzylisoquinoline alkaloids [28]. The integration of AI tools is expected to accelerate the identification of novel biosynthetic pathways from the vast amount of available genomic and metabolomic data.
Single-Cell and Spatial Omics
Emerging techniques such as single-cell sequencing and MS imaging enable researchers to probe metabolic processes at unprecedented resolution, revealing the spatial organization of pathways within specific cell types [28] [29]. This is particularly important for plant natural products, which are often produced in highly specific cell types or organelles. Recent high-resolution analyses at the level of specific cell types, individual cells, or even organelles have revealed remarkable compartmentalization of plant metabolic pathways [28].
Sustainable Production through Metabolic Engineering
With complete biosynthetic pathways in hand, researchers are increasingly focusing on metabolic engineering strategies for sustainable production of valuable natural products. This includes engineering microbial hosts like yeast or bacteria to produce plant natural products, as demonstrated for artemisinic acid [29], as well as optimizing plant cell cultures for enhanced production of target compounds. Future directions include metabolon engineering, AI integration, and developing cheaper and greener production strategies for plant natural products [4].
The evolution of natural product discovery from simple plant extracts to comprehensive pathway elucidation represents a remarkable scientific journey that has transformed our understanding of plant chemical diversity. This transition has been enabled by the integration of multi-omics technologies, advanced computational tools, and innovative experimental approaches that collectively illuminate how plants transform simple primary metabolites into complex chemical scaffolds with significant pharmacological activities.
The ongoing integration of artificial intelligence, single-cell technologies, and sophisticated engineering approaches promises to further accelerate this field, potentially unlocking the full therapeutic potential of plant natural products while enabling sustainable production systems. As these advancements continue, our ability to decode and harness nature's chemical ingenuity will undoubtedly lead to new therapeutic agents and deeper insights into the fundamental biochemical principles that govern natural product biosynthesis.
Engineering Nature's Pathways: Methodologies for Harnessing and Applying Biosynthetic Logic
The field of metabolic engineering has undergone a significant transformation, entering a third wave characterized by the application of synthetic biology principles to design and construct complete metabolic pathways in microbial hosts for the production of noninherent chemicals [30]. This approach enables the systematic engineering of microbes such as E. coli and yeast to function as efficient cell factories, converting renewable biomass into valuable chemicals, fuels, and pharmaceuticals [31] [30]. Pathway reconstruction involves the careful selection of genetic parts, their assembly into functional pathways, and the optimization of metabolic flux to achieve high titers, yields, and productivity of target compounds [31]. This technical guide outlines the core principles and methodologies for successful heterologous pathway expression, framed within the context of producing biosynthetic building blocks from primary metabolism.
The synthetic biology approach to metabolic engineering typically follows an iterative workflow comprising four key stages: design, modeling, synthesis, and analysis [31]. This framework provides a standardized methodology for building biological systems from well-characterized, modular parts, moving beyond the traditional trial-and-error approach to a more predictable, engineering-based discipline [31]. The application of this framework is particularly valuable for rewiring cellular metabolism to enhance the production of target compounds, including medicinal plant bioactive compounds, where challenges such as long metabolic pathways, inadequate catalytic efficiency of key enzymes, and incompatibility between genetic elements and host cells often limit yields [32].
Core Principles and Design Framework
The Four-Stage Engineering Cycle
The forward engineering of synthetic metabolic pathways relies on a cyclical process that integrates computational design with experimental validation [31].
Design: This initial phase involves selecting appropriate genetic parts and formulating a blueprint for the metabolic pathway. The design process requires explicit specification of each necessary component, including promoters, ribosomal binding sites (RBSs), protein-coding sequences, and terminators [31]. At the pathway level, design focuses on mixing and matching modular parts while implementing control mechanisms to balance and optimize metabolic flux [31].
Modeling: Computational models are employed to predict system behavior before physical construction. Model-guided design approaches limit system variability by fitting mathematical models with measured parameters, increasing predictability and decreasing time spent on combinatorial system construction, testing, and debugging [31]. Genome-scale metabolic models are particularly valuable for exploring the metabolic potential of cell factories and identifying target genes for engineering [30].
Synthesis: This stage involves the physical construction of the designed genetic system using recombinant DNA technology. Advances in de novo DNA synthesis and codon optimization contribute significantly to manufacturing pathway enzymes with improved or novel function [31]. Standardized assembly methods, such as the BioBrick methodology, facilitate the construction process through well-defined genetic parts [31].
Analysis: The constructed pathways are experimentally validated through rigorous analysis of performance metrics. Analytical methods assess pathway functionality, metabolic flux, and product formation, generating data that inform subsequent design iterations [31]. This stage provides critical feedback for refining the system and improving its performance.
The following diagram illustrates this iterative engineering workflow:
Hierarchical Engineering Strategies
Modern metabolic engineering operates across multiple biological hierarchies to systematically rewire cellular metabolism [30]. This hierarchical approach enables precise intervention at different levels of cellular organization:
Part Level: Engineering focuses on individual genetic components, including promoters, RBSs, protein-coding sequences, and terminators. Libraries of standardized, characterized parts facilitate the predictable design of genetic circuits [31]. Key considerations at this level include codon optimization to match host preferences and the elimination of restriction sites for standardized assembly [31].
Pathway Level: Engineering involves the assembly of multiple genetic parts into functional metabolic pathways. At this level, balancing metabolic flux through tunable control mechanisms becomes critical [31]. Strategies include enzyme colocalization using protein scaffolds that bear modular interaction domains to physically link pathway enzymes [31].
Network Level: Engineering considers the interaction between the heterologous pathway and the host's native metabolic network. This may involve deleting competing pathways, overexpressing bottleneck enzymes, or modulating regulatory networks to redirect flux toward the desired product [30].
Genome Level: Engineering employs genome editing techniques to implement system-wide modifications. This includes creating knockout strains, integrating heterologous genes at specific genomic locations, and implementing genome-scale changes to optimize host performance [30].
Cell Level: Engineering addresses cellular properties beyond metabolism, including growth characteristics, stress tolerance, and product secretion. This may involve engineering transporter proteins to enhance substrate uptake or product efflux, or modifying cellular machinery to improve tolerance to toxic intermediates or products [30].
The relationship between these hierarchical levels is visualized in the following diagram:
Experimental Protocols and Methodologies
Pathway Design and Codon Optimization
The successful reconstruction of heterologous pathways begins with careful in silico design and optimization of genetic components [31].
Codon Optimization: Protein-coding sequences must be optimized for expression in the heterologous host. Codon usage bias can significantly impact expression levels, and suboptimal codon usage may result in poor enzyme expression or misfolded proteins [31]. Utilize freely available algorithms such as Gene Designer or similar tools to encode the same amino acid sequence with alternative, preferred nucleotide sequences that match the host's codon preference [31].
Standardized Part Design: Genetic parts should comply with standard assembly requirements, such as the exclusion of specific restriction enzyme sites reserved for assembly in methodologies like BioBricks [31]. Additionally, part-specific objectives including activity or specificity modifications should be considered during the design phase [31].
Regulatory Element Selection: Choose appropriate promoters, RBSs, and terminators based on desired expression levels. Well-characterized part libraries, such as constitutive promoter libraries with varying strengths, enable fine-tuning of gene expression [31]. For inducible systems, select regulator-operator pairs that minimize cross-talk with host systems [31].
Assembly and Transformation
The physical construction of metabolic pathways involves the assembly of genetic parts and their introduction into the host chassis [31].
DNA Assembly: Utilize standardized assembly methods such as BioBricks, Golden Gate, or Gibson assembly to combine genetic parts into functional pathways. The choice of method depends on the number of parts, available resources, and compatibility with existing part libraries [31]. Ensure all parts are compatible with the selected assembly standard.
Host Transformation: Introduce the assembled genetic constructs into the microbial chassis using appropriate transformation methods. For E. coli, heat shock or electroporation are commonly used, while yeast typically requires lithium acetate or electroporation methods. Selectable markers are essential for identifying successful transformants.
Vector Selection: Choose appropriate vectors based on copy number, compatibility with the host, and stability. Origins of replication significantly impact plasmid copy number and should be selected based on desired expression levels [31]. For metabolic engineering applications, consider using low-copy vectors to reduce metabolic burden on the host.
Screening and Analysis
Following transformation, rigorous screening and analysis are required to identify successful pathway reconstruction and functionality [31].
Functional Screening: Implement high-throughput screening methods to identify clones with desired metabolic activity. This may include colorimetric assays, growth-based selection, or analytical techniques such as HPLC or GC-MS to detect product formation [33].
Pathway Analysis Tools: Utilize computational tools to analyze the performance of reconstructed pathways. Over-representation analysis and pathway topology analysis can help determine whether certain pathways are enriched in the engineered strains [34]. Tools such as Reactome provide statistical tests to identify over-represented pathways and visualize how submitted identifiers map to known pathways [34].
Flux Analysis: Employ metabolic flux analysis to quantify the flow of metabolites through pathways and identify potential bottlenecks. Techniques such as 13C-labeling can provide insights into intracellular flux distributions and guide further engineering efforts [30].
Case Studies and Performance Metrics
Representative Production Metrics in Engineered Hosts
The table below summarizes performance metrics for various chemicals produced through heterologous pathway expression in microbial chassis, demonstrating the effectiveness of hierarchical metabolic engineering strategies [30].
| Chemical | Host | Titer (g/L) | Yield (g/g) | Productivity (g/L/h) | Key Engineering Strategies |
|---|---|---|---|---|---|
| L-Lactic Acid | C. glutamicum | 212 | 0.98 | - | Modular pathway engineering [30] |
| Succinic Acid | E. coli | 153.36 | - | 2.13 | Modular pathway engineering, High-throughput genome engineering, Codon optimization [30] |
| 3-Hydroxypropionic Acid | C. glutamicum | 62.6 | 0.51 | - | Substrate engineering, Genome editing engineering [30] |
| Lysine | C. glutamicum | 223.4 | 0.68 | - | Cofactor engineering, Transporter engineering, Promoter engineering [30] |
| Muconic Acid | C. glutamicum | 54 | 0.20 | 0.34 | Modular pathway engineering, Chassis engineering [30] |
| Malonic Acid | Y. lipolytica | 63.6 | - | 0.41 | Modular pathway engineering, Genome editing engineering, Substrate engineering [30] |
| Valine | E. coli | 59 | 0.39 | - | Transcription factor engineering, Cofactor engineering, Genome editing engineering [30] |
Advanced Engineering Applications
Beyond standard pathway reconstruction, several advanced applications demonstrate the cutting edge of heterologous expression technology:
Artemisinin Production: The complete metabolic pathway for artemisinic acid, a precursor to the antimalarial drug artemisinin, was reconstructed in yeast through extensive engineering of the mevalonate pathway and amorphadiene synthesis, followed by oxidation to artemisinic acid [30]. This landmark achievement demonstrated the potential for microbial production of complex plant-derived pharmaceuticals.
Enzyme Colocalization: Inspired by natural systems, protein scaffolds bearing modular interaction domains can physically link pathway enzymes tagged with corresponding peptide ligands [31]. This elegant approach enhances pathway efficiency by promoting substrate channeling and reducing intermediate diffusion.
RNA Devices: Synthetic RNA devices incorporating aptamers for sensing small molecules, transmitter sequences, and actuator elements such as ribozymes can provide sophisticated regulation of metabolic pathways [31]. These devices enable dynamic control of pathway expression in response to metabolic status.
The Scientist's Toolkit: Essential Research Reagents
Successful pathway reconstruction requires a comprehensive toolkit of genetic parts, analytical tools, and computational resources. The table below details essential research reagents and their applications in heterologous pathway engineering [31] [34] [33].
| Research Tool | Function and Application | Key Features |
|---|---|---|
| Standard Biological Parts | Modular genetic elements for pathway construction [31] | Promoters, RBSs, coding sequences, terminators; Standardized for interoperability |
| Codon Optimization Software | Algorithmic optimization of coding sequences for heterologous hosts [31] | Adapts codon usage to host preferences; Tools: Gene Designer, DNA2.0 |
| Registry of Standard Biological Parts | Repository of characterized genetic parts [31] | Collection of standardized, reusable biological components |
| Pathway Analysis Tools | Computational identification of enriched pathways in engineered strains [34] [33] | Tools: g:Profiler, GSEA, Reactome; Statistical over-representation analysis |
| Genome-Scale Metabolic Models | Computational models predicting metabolic fluxes [30] | Identifies gene knockout/overexpression targets; Platforms: COBRA tools |
| RNA Devices | Post-transcriptional regulation of pathway expression [31] | Aptamer sensors, transmitter sequences, ribozyme actuators; Dynamic control |
| Protein Scaffolds | Physical colocalization of pathway enzymes [31] | Modular interaction domains with peptide ligands; Enhances metabolic channeling |
| L-Cysteine ethyl ester HCl | L-Cysteine Ethyl Ester Hydrochloride|RUO | Research-grade L-Cysteine ethyl ester hydrochloride for studying opioid side effects, antioxidant mechanisms, and more. For Research Use Only. Not for human use. |
| O-tert-Butylthreoninetert-butyl ester | (2S,3R)-tert-Butyl 2-amino-3-(tert-butoxy)butanoate | Explore (2S,3R)-tert-Butyl 2-amino-3-(tert-butoxy)butanoate for life science research. This compound is a key building block in organic synthesis. For Research Use Only. Not for human use. |
Pathway reconstruction for heterologous expression in microbial chassis represents a powerful paradigm for the sustainable production of valuable chemicals from renewable resources. The synthetic biology approach, with its emphasis on design, modeling, synthesis, and analysis, provides a rigorous framework for engineering microbial cell factories [31]. Hierarchical strategies that intervene at the part, pathway, network, genome, and cell levels enable comprehensive rewiring of cellular metabolism to optimize production metrics [30].
Future advancements in the field will likely focus on the development of more sophisticated regulatory tools, enhanced computational models for predicting pathway performance, and improved genome editing technologies for rapid strain optimization [32] [30]. The integration of machine learning approaches for designing optimal genetic constructs and predicting metabolic fluxes holds particular promise for accelerating the design-build-test cycle [30]. As these technologies mature, heterologous pathway expression in microbial chassis will play an increasingly important role in the sustainable production of chemicals, fuels, and pharmaceuticals, ultimately reducing resource consumption and environmental impact associated with traditional production methods [32].
Combinatorial biosynthesis is a powerful genetic engineering strategy that expands the biosynthetic inventory of native producers by introducing non-native enzymes into specific pathways, thereby manipulating natural product output to generate structurally diversified molecules [35]. This approach represents a fusion of genetic engineering and natural product chemistry, allowing researchers to extend nature's biosynthetic dexterity by reprogramming natural pathways through the mixing and matching of genes from known biosynthetic clusters [36]. The fundamental motivation driving the field is the production of "unnatural" natural products with altered structures that can illuminate structure-activity relationships crucial for drug development while improving the pharmaceutical properties of clinically relevant compounds [36].
This technical guide frames combinatorial biosynthesis within the broader context of primary metabolism research, wherein simple building blocks from central metabolic pathways—such as acyl-CoAs from fatty acid metabolism, amino acids from protein synthesis, and isopentenyl pyrophosphate from the mevalonate pathway—serve as the foundational substrates for engineered biosynthetic systems [35]. By harnessing and redirecting the flux of these primary metabolic building blocks through engineered pathways, researchers can create novel chemical entities that expand the accessible chemical space for drug discovery and development.
Foundational Principles and Building Blocks
Biosynthetic Origins of Natural Product Scaffolds
Natural products are classified according to their biosynthetic origin, with major classes including polyketides, non-ribosomal peptides, terpenes, and hybrid molecules that combine structural elements from multiple pathways [35]. From a biosynthetic perspective, the diversity and complexity of natural products are generated through a two-step process: (1) formation of the core hydrocarbon scaffold by megasynth(et)ases, and (2) modification of this scaffold by tailoring enzymes [35].
Fungal natural products, in particular, are produced via highly programmed pathways originating from simple building blocks derived from primary metabolism, including acyl-CoAs, proteinogenic and non-proteinogenic amino acids, isopentenyl pyrophosphate (IPP)/dimethylallylpyrophosphate (DMAPP), and sugars [35]. The engineered rerouting of these universal building blocks provides the foundation for combinatorial biosynthesis approaches.
Molecular Complexity Metrics for Pathway Evaluation
Recent advances in informatic methodology have enabled systematic comparison between biological and chemical synthetic strategies using molecular complexity metrics [37]. Key descriptors include:
- Molecular Weight (MW): Measured in Daltons (Da)
- Fraction of sp3 hybridized carbon atoms (Fsp3): Indicator of three-dimensionality
- Complexity Index (Cm): Quantitative measure of structural complexity
These metrics can be visualized in 3D plots parameterized by Fsp3, Cm, and MW to observe how complexity changes throughout a synthetic pathway, with efficient pathways creating complex specialized metabolites in as few processes as possible [37]. This analytical framework allows researchers to quantitatively compare the efficiency of combinatorial biosynthesis approaches against traditional chemical synthesis routes.
Key Engineering Strategies in Combinatorial Biosynthesis
Megasynth(et)ase Engineering
Megasynth(et)ases are large, multifunctional enzymes that synthesize the essential carbon framework of natural products. For polyketide synthases (PKSs), particularly non-reducing PKSs (NR-PKSs), several domain swapping strategies have been successfully employed:
Table: Domain Swapping Strategies in Non-Reducing PKS (NR-PKS) Engineering
| Domain Type | Function | Engineering Approach | Result |
|---|---|---|---|
| Starter Unit Acyl Carrier Protein Transacylase (SAT) | Selects and transfers starter unit to ketosynthase domain [35] | Swapping between AfoE and StcA | Novel polyketide utilizing hexanoyl starter unit [35] |
| Product Template (PT) | Essential for cyclization and aromatization of polyketide chain [35] | Swap of PT from ApdA into PKS4 | Production of novel α-pyranoanthraquinone [35] |
| C-Methyltransferase (CMeT) | Catalyzes methylation of growing polyketide chain [35] | Combinatorial swaps between multiple NR-PKSs | Revealed kinetic competition with KS domain may override CMeT function [35] |
| Thiolesterase (TE) | Catalyzes polyketide cyclization and release [35] | Swapping between AtCURS1/2 and CcRADS1/2 | Generated multiple macrocycles, pyrones, carboxylic acids, and esters [35] |
For highly reducing PKS (HR-PKS), engineering challenges increase due to the frequent absence of terminal release domains and difficulties in detecting non-aromatic products [35]. Successful examples include enoylreductase (ER) domain swaps in DrtA, the HR-PKS involved in biosynthesis of fungal drimane-type sesquiterpene esters, which led to production of novel metabolites including calidoustrene F with different levels of saturation in the attached polyketide chain [35].
Building Block Pathway Engineering
Structural diversification by combinatorial biosynthesis can be limited by the substrate specificity of biosynthetic enzymes. Key engineering approaches include:
Expanding Polyketide Extender Unit Repertoire
The gatekeeper enzyme domain in modular PKSs is the acyltransferase (AT) domain that controls selection and incorporation of extender units (usually malonyl-, methylmalonyl-, or ethylmalonyl-CoAs) [36]. The restricted versatility of polyketide extender units has historically limited generation of novel polyketide structures, but this constraint has been addressed through:
- Discovery of crotonyl-CoA carboxylase/reductase (CCR) enzymes that catalyze reductive carboxylation of α,β-unsaturated acyl-CoA precursors [36]
- Engineering of AT domain specificity through amino acid substitutions to alter extender unit selectivity [36]
- Utilization of malonyl-CoA synthetase (MatB) with naturally or engineered promiscuous AT domains to incorporate diverse extender units [36]
For example, the AT domain of module 4 in the immunosuppressant FK506 PKS naturally accepts methylmalonyl-, ethylmalonyl-, propylmalonyl-, and allylmalonyl-CoA substrates as well as unnatural acyl-CoAs, generating macrolide derivatives with modified C21 side chains, one of which exhibited improved in vitro nerve regenerative activity relative to the parent FK506 [36].
Reprogramming Non-Ribosomal Peptide Assembly
In non-ribosomal peptide synthetase (NRPS) systems, adenylation (A) domains control the entry of diverse amino acid substrates to the NRPS assembly line. Engineering strategies include:
- Rational design through point mutations in specificity-determining residues
- Directed evolution of A domains through saturation mutagenesis of specificity-conferring sites
- Exploitation of natural substrate promiscuity in selected A domains
A notable example includes the modification of the A domain of module 10 within the calcium-dependent antibiotic (CDA) NRPS through a single mutation (Lys278Gln), changing its specificity from (2S,3R)-3-methyl Glu (mGlu)/Glu to (2S,3R)-3-methyl Gln (mGln)/Gln to produce novel CDA analogues [36].
Experimental Workflows and Methodologies
Heterologous Pathway Expression Systems
The implementation of combinatorial biosynthesis strategies typically requires reconstruction of engineered pathways in suitable heterologous hosts. Well-established experimental workflows include:
4.1.1 Fungal Host Engineering in Aspergillus oryzae
- Protocol: The biosynthetic pathway for sporothriolide was fully reconstructed in Aspergillus oryzae, requiring expression of genes encoding two fungal fatty acid synthase components (SpofasA & B), an alkyl citrate synthase (SpoE), a methylcitrate dehydratase homolog (SpoL), a decarboxylase (SpoK), a non-heme iron dioxygenase (SpoG), and hydrolases SpoH and SpoJ [37].
- Key Considerations: Use of strong inducible promoters, codon optimization for heterologous expression, and balanced expression of large multimodular proteins.
4.1.2 Bacterial Host Engineering in Escherichia coli
- Protocol: Engineering of E. coli for production of novel polyketides through expression of heterologous PKS genes, including those requiring specific cofactors or post-translational modifications.
- Key Considerations: Implementation of phosphopantetheinyl transferases for ACP activation, co-expression of precursor supply pathways, and optimization of fermentation conditions.
DNA Assembly and Pathway Refactoring
Modern combinatorial biosynthesis relies on advanced DNA assembly techniques for pathway construction:
- Golden Gate Assembly: For modular assembly of large biosynthetic gene clusters
- Yeast Assembly: For reconstruction of very large gene clusters (>50 kb)
- CRISPR-Cas Mediated Genome Editing: For precise genome integration of engineered pathways
- Modular Cloning Systems: Such as MoClo or GoldenBraid for standardized parts assembly
Table: Key Research Reagent Solutions for Combinatorial Biosynthesis
| Reagent/Resource | Function | Application Examples |
|---|---|---|
| Aspergillus oryzae heterologous expression system | Robust fungal host for expression of fungal biosynthetic pathways [37] | Reconstruction of sporothriolide biosynthetic pathway [37] |
| Escherichia coli BAP1 strain | Bacterial host engineered for heterologous expression of PKS and NRPS pathways | Production of novel polyketides and non-ribosomal peptides |
| Type IIS restriction enzymes (BsaI, BsmBI) | Enable Golden Gate assembly of genetic parts | Modular construction of engineered biosynthetic pathways |
| CRISPR-Cas9 systems for fungal and bacterial engineering | Precision genome editing tool | Gene knockouts, promoter replacements, and pathway integrations |
| Phosphopantetheinyl transferases (Sfp, NpgA) | Activate carrier proteins in PKS and NRPS systems | Essential for functionality of heterologously expressed megasynth(et)ases |
| Crotonyl-CoA carboxylase/reductase (CCR) enzymes | Generate diverse extender units for PKS engineering [36] | Expansion of polyketide structural diversity through unnatural extender units |
Analytical and Computational Tools
Cheminformatic Approaches for Natural Product Analysis
The structural complexity of engineered natural products necessitates advanced analytical and computational tools:
5.1.1 Biosynfoni Molecular Fingerprint
Biosynfoni is a natural product-specific molecular fingerprint based on a relatively small set of 39 selected biosynthetic building blocks that provides more interpretable predictions of biosynthetic distance and natural product classification [38]. Key features include:
- Biosynthetic Relevance: Structural features can be directly linked to the biosynthesis of the molecule
- Enhanced Interpretability: Values directly represent substructures as opposed to hashed fingerprints
- Computational Efficiency: Faster generation than MACCS and RDKit fingerprints despite pure-Python coding [38]
Biosynfoni captures biosynthetic changes along biosynthetic reaction chains, showing a continuous decrease in similarity scores as more reactions separate compound pairs, unlike traditional fingerprints [38].
5.1.2 Metabolic Pathway Visualization and Analysis
Tools for visualizing engineered metabolic pathways include:
- antiSMASH: For identification and analysis of biosynthetic gene clusters
- MIBiG: Minimum Information about a Biosynthetic Gene Cluster repository
- BiG-SCAPE: For comparative analysis of biosynthetic gene clusters
Applications and Future Perspectives
Pharmaceutical Applications
Combinatorial biosynthesis has yielded numerous compounds with optimized pharmaceutical properties:
- Spinetoram: A spinosyn-based insecticide developed through a combination of biological and chemical approaches, marketed in 2007 with improved efficacy and expanded spectrum [36]
- Novel daptomycin analogues: Over 120 novel lipopeptides generated through combinatorial biosynthesis, some displaying improved therapeutic properties [36]
- FK506 analogues: Macrolide derivatives with modified C21 side chains, one exhibiting improved in vitro nerve regenerative activity [36]
Integration with Synthetic Biology and AI
Future directions in combinatorial biosynthesis include:
- AI-Integrated Pathway Design: Machine learning approaches for predicting enzyme compatibility and pathway efficiency
- Automated Strain Engineering: High-throughput robotic systems for pathway assembly and screening
- Biosensor-Enabled Screening: Implementation of genetically encoded biosensors for rapid detection of desired compounds
- Cell-Free Biosynthetic Systems: Platforms for rapid prototyping of engineered pathways without cellular constraints
The continued development of combinatorial biosynthesis approaches promises to significantly expand the chemical space accessible for drug discovery and development, building upon nature's biosynthetic logic while expanding the structural diversity of biologically active natural products.
Enzyme promiscuity, the inherent ability of enzymes to catalyze secondary reactions alongside their native functions, has emerged as a cornerstone for engineering novel biosynthetic pathways. This whitepaper provides an in-depth technical guide on harnessing and enhancing this property for the transformation of non-natural substrates. Framed within the context of primary metabolism research, we detail the mechanistic basis of promiscuity, present quantitative frameworks for its assessment, and outline robust experimental protocols for its directed evolution. A particular emphasis is placed on leveraging promiscuous activities to generate non-canonical biosynthetic building blocks. This guide is intended to equip researchers and drug development professionals with the advanced methodologies needed to expand the synthetic biology toolkit for the production of high-value chemicals and pharmaceuticals.
Enzyme promiscuity is broadly defined as the capacity of an enzyme to catalyze either a comparable chemical transformation on different substrates (substrate promiscuity) or an entirely different type of chemical reaction (catalytic promiscuity) [39]. Historically, enzymes involved in central, or primary, metabolism were thought to be exemplars of specificity. However, a growing body of evidence reveals that these enzymes are often remarkably versatile. It is now estimated that ~37% of metabolic enzymes in E. coli catalyze promiscuous reactions, affecting at least 65% of metabolic reactions [40]. This "underground metabolism" is not merely a biochemical curiosity; it is a fundamental feature that provides metabolic networks with robustness, resilience, and a built-in capacity for evolutionary innovation [40] [41].
From an engineering perspective, promiscuity is the key that unlocks the potential to repurpose primary metabolism. The enzymes of central metabolism have already been optimized by evolution to handle core cellular metabolites—such as pyruvate, acetyl-CoA, and glyoxylate—with high proficiency. Their inherent flexibility allows them to accept non-natural analogues of these metabolites, making them ideal starting points for engineering novel pathways that branch from central metabolic nodes [41]. This strategy is paramount for the sustainable production of biosynthetic building blocks for pharmaceuticals, polymers, and fine chemicals, moving beyond the limited repertoire of natural products.
Quantitative Assessment of Promiscuous Activities
A critical first step in engineering promiscuity is the accurate quantification of an enzyme's native and non-native activities. The catalytic efficiency (k_cat/K_M) is the gold standard for this assessment, as it reflects the enzyme's overall ability to convert a substrate to a product.
Key Kinetic Parameters
The following table summarizes the typical range of catalytic efficiencies for native versus promiscuous activities, illustrating the "efficiency gap" that engineering must overcome.
Table 1: Characteristic Kinetic Parameters of Enzyme Activities
| Activity Type | Typical kcat/KM Range (Mâ»Â¹sâ»Â¹) | Example Enzyme | Example Substrate |
|---|---|---|---|
| Native (Primary) | 10ⵠ- 10⸠| SerB (E. coli phosphatase) | Phosphoserine [42] |
| Promiscuous (Strong) | 10â´ - 10âµ | cN-IIIB (Human phosphatase) | m7GMP [40] |
| Promiscuous (Weak) | 10â»Â¹ - 10³ | HisB, Gph, YtjC (E. coli phosphatases) | Phosphoserine [42] |
High-Throughput Screening for Promiscuity
Broad-scale profiling of enzyme families has revealed the vast potential of promiscuous activity space. For instance, a screen of 217 members of the haloacid dehalogenase (HAD) family against 169 phosphorylated compounds found that over 90% of the enzymes hydrolyzed a median of 15.5 substrates, with some acting on over 140 [39]. Such studies provide rich datasets for identifying promising engineering starting points.
Experimental Protocols for Harnessing and Enhancing Promiscuity
This section outlines core methodologies for discovering, quantifying, and evolving promiscuous activities.
Protocol 1: Construction and Use of a Biosensor Strain for Pathway Discovery
Principle: Synthetic auxotrophic strains, lacking one or more genes in an essential biosynthetic pathway, can be used to select for promiscuous activities that bypass the metabolic lesion [41].
Application Example: Discovering a recursive isoleucine biosynthesis pathway in E. coli [41].
Strain Engineering:
- Start with a base E. coli K-12 strain (e.g., MG1655).
- Sequentially delete genes encoding all known threonine deaminases (
ilvA,tdcB,sdaA,sdaB,tdcG) to create a strain auxotrophic for 2-ketobutyrate (2KB), the precursor to isoleucine. - Optionally, delete the
metBgene (cystathionine γ-synthase) to eliminate a known underground route to 2KB, creating a more stringent Isoleucine-Methionine auxotroph (IMaux).
Selection and Evolution:
- Grow the IMaux strain in M9 minimal medium with glucose or glycerol as a carbon source, supplemented with methionine but lacking isoleucine/2KB.
- Passage the culture repeatedly to allow for spontaneous mutations that restore growth.
- Isolate evolved clones capable of robust growth without isoleucine supplementation.
Pathway Identification:
- Sequence the genomes of evolved clones to identify causal mutations. The reactivation of a frameshifted
ilvGgene (encoding acetohydroxyacid synthase II, AHAS II) is a key finding. - Confirm the recursive pathway by
¹³Cisotopic labeling of potential precursors (e.g., glyoxylate and pyruvate) and track the incorporation of label into 2KB and isoleucine using GC- or LC-MS.
- Sequence the genomes of evolved clones to identify causal mutations. The reactivation of a frameshifted
Diagram 1: Biosensor strain workflow for discovering promiscuous pathways that produce essential metabolites like isoleucine.
Protocol 2: Directed Evolution of a Promiscuous Activity
Principle: By applying selective pressure for a desired, initially inefficient promiscuous activity, one can dramatically improve its catalytic efficiency through iterative rounds of mutagenesis and screening [43].
Application Example: Evolving a phosphotriesterase (PTE) from Pseudomonas diminuta to become an efficient arylesterase [43].
Library Creation:
- Subject the gene encoding the wild-type PTE to random mutagenesis (e.g., error-prone PCR) or gene shuffling to create a library of variants.
High-Throughput Screening:
- Express the variant library in a suitable host (e.g., E. coli).
- Plate colonies on solid medium or grow in microtiter plates containing a chromogenic or fluorogenic aryl ester substrate (e.g., p-nitrophenyl acetate).
- Identify clones that exhibit increased hydrolysis of the aryl ester substrate relative to the wild-type PTE.
Iteration and Characterization:
- Isolate the best-performing variants. Use them as templates for further rounds of mutagenesis and screening.
- After multiple rounds (e.g., 18), purify the evolved enzyme and determine its kinetic parameters (
k_cat,K_M) for both the native (organophosphate) and new (aryl ester) substrates. The result is often a significant shift in specificity, sometimes by a factor of 10â¹ [43].
The Scientist's Toolkit: Essential Research Reagents and Materials
Success in engineering enzyme promiscuity relies on a specific set of biological and chemical reagents.
Table 2: Key Research Reagent Solutions for Enzyme Promiscuity Engineering
| Reagent / Material | Function and Rationale | Example Use Case |
|---|---|---|
| Biosensor Strains | Engineered microbial strains auxotrophic for a specific metabolite; used for selecting/enhancing promiscuous activities that produce the missing metabolite. | E. coli Δ5 (ΔilvA, ΔtdcB, etc.) for discovering novel 2KB/Isoleucine pathways [41]. |
| Structured Substrate Libraries | A diverse collection of potential substrates (e.g., 169 phosphorylated metabolites); enables high-throughput profiling of substrate ambiguity. | Defining the substrate specificity profile of HAD superfamily phosphatases [40] [39]. |
| Gene Mutagenesis Kits | Kits for error-prone PCR or DNA shuffling; essential for creating genetic diversity as the starting point for directed evolution. | Generating variant libraries of phosphotriesterase (PTE) for directed evolution [43]. |
| Chromogenic/Fluorogenic Substrate Probes | Synthetic substrates that produce a measurable signal (color or fluorescence) upon enzyme action; crucial for high-throughput screening. | Screening PTE variant libraries for improved arylesterase activity using p-nitrophenyl acetate [43]. |
| Ancestral Sequence Reconstruction (ASR) Tools | Computational and synthetic biology tools to infer and synthesize ancestral enzymes; can reveal generalist catalysts with broader promiscuity. | Studying the evolution of specificity in mammalian immune proteases or vertebrate steroid receptors [43]. |
| H-DL-Phe(4-NO2)-OH | H-DL-Phe(4-NO2)-OH, CAS:2922-40-9, MF:C9H10N2O4, MW:210.19 g/mol | Chemical Reagent |
| Cyclohexylglycine | Cyclohexylglycine, CAS:14328-51-9, MF:C8H15NO2, MW:157.21 g/mol | Chemical Reagent |
Visualization of Engineering Strategies and Metabolic Outcomes
Engineering promiscuity often involves creating new metabolic pathways that tap into central metabolism. The recursive isoleucine pathway discovered in E. coli provides an excellent example of this principle.
Diagram 2: A recursive pathway for isoleucine biosynthesis. The promiscuous activity of AHASII on glyoxylate and pyruvate generates 2KB, which is then used recursively by the same enzyme in a canonical reaction with another pyruvate to initiate isoleucine synthesis.
The deliberate engineering of enzyme promiscuity represents a paradigm shift in metabolic engineering and synthetic biology. By viewing the inherent "sloppiness" of enzymes not as a flaw but as a feature, researchers can access a vast landscape of novel chemistry directly from central metabolism. The protocols and strategies outlined herein—from using clever biosensor strains for in vivo evolution to high-throughput in vitro screening and the application of advanced computational models like EPP-HMCNF for activity prediction [44]—provide a robust framework for this endeavor.
Future advances will be driven by an even deeper integration of computational and experimental approaches. Machine learning models, trained on the ever-growing databases of enzyme kinetics and structures, will become indispensable for predicting promising enzyme-substrate pairs and guiding mutagenesis strategies [45] [44]. Furthermore, a better understanding of biophysical constraints and "frustration" —where competing interactions limit enzyme specialization—will help design more effective evolutionary trajectories [46]. As we continue to illuminate the intricate connections between primary and underground metabolism, the toolkit for creating bespoke biosynthetic pathways will expand, accelerating the development of bio-based manufacturing and drug discovery.
Artemisinin, a sesquiterpene lactone containing a crucial endoperoxide bridge, stands as the most potent antimalarial drug currently available [47]. This natural product is synthesized and stored in the glandular secretory trichomes (GSTs) of the plant Artemisia annua [48] [47]. The discovery of artemisinin by Professor Youyou Tu, awarded the Nobel Prize in 2015, and its subsequent development into Artemisinin-based Combination Therapies (ACTs) has revolutionized malaria treatment, saving countless lives worldwide [47] [49]. ACTs are the World Health Organization (WHO)-recommended first-line treatment for uncomplicated P. falciparum malaria [50] [49].
Despite its efficacy, the natural production of artemisinin faces significant challenges. The artemisinin content in wild-type A. annua is low, typically ranging from 0.1% to 1.0% of plant dry weight, making large-scale extraction resource-intensive and costly [51] [47]. Furthermore, the agricultural supply chain is susceptible to seasonal and price fluctuations, leading to periods of both shortage and oversupply [52]. The complex chemical structure of artemisinin, featuring the unique endoperoxide bridge, makes its total chemical synthesis economically unviable for large-scale production [51] [53].
To address these challenges and create a stable, scalable second source of artemisinin, a semi-synthetic production platform was developed. This approach ingeniously leverages biosynthetic building blocks from primary metabolism in an engineered microbial host, followed by a chemical conversion to the final product. This case study details the technical development of this successful semi-synthetic production process, from the engineering of microbial chassis to the final chemical synthesis, framing it within the context of harnessing primary metabolism for the production of a high-value secondary metabolite.
The Biosynthetic Pathway and Engineering Rationale
Artemisinin Biosynthesis inArtemisia annua
A comprehensive understanding of the native biosynthetic pathway in A. annua is fundamental to recreating it in a heterologous host. Artemisinin is a sesquiterpene, deriving from the universal five-carbon isoprenoid precursors, isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) [47] [53]. These building blocks are supplied by two primary metabolic pathways: the cytosolic mevalonate (MVA) pathway and the plastidial methylerythritol phosphate (MEP) pathway [47]. In A. annua, the cytosolic MVA pathway primarily provides the flux for sesquiterpene biosynthesis [47].
The committed pathway to artemisinin begins with the condensation of three IPP units (derived from acetyl-CoA) to form the C15 intermediate farnesyl pyrophosphate (FPP). The biosynthesis then proceeds through a series of specialized, cytochrome P450-mediated oxidation steps, as detailed below and in Figure 1 [52] [47] [53].
Table 1: Key Enzymes in the Artemisinin Biosynthetic Pathway.
| Enzyme | Abbreviation | Function in Pathway |
|---|---|---|
| Amorpha-4,11-diene Synthase | ADS | Cyclizes FPP to form amorpha-4,11-diene, the first dedicated step. |
| Cytochrome P450 Monooxygenase | CYP71AV1 | Multi-functional oxidase; hydroxylates amorpha-4,11-diene to artemisinic alcohol. |
| Cytochrome P450 Reductase | CPR | Redox partner for CYP71AV1, supplies electrons. |
| Alcohol Dehydrogenase 1 | ADH1 | Oxidizes artemisinic alcohol to artemisinic aldehyde. |
| Aldehyde Dehydrogenase 1 | ALDH1 | Oxidizes artemisinic aldehyde to artemisinic acid (AA). |
| Double Bond Reductase 2 | DBR2 | Reduces artemisinic aldehyde to dihydroartemisinic aldehyde (branch point). |
| Aldehyde Dehydrogenase 1 (also) | ALDH1 | Oxidizes dihydroartemisinic aldehyde to dihydroartemisinic acid (DHAA). |
The final conversion of the precursor dihydroartemisinic acid (DHAA) to artemisinin occurs spontaneously via a non-enzymatic photo-oxidation reaction [47] [53]. Artemisinic acid (AA) was identified as a stable, high-yield precursor that could be produced in a microbial host and then efficiently converted to artemisinin through a defined chemical process, forming the basis of the semi-synthetic strategy [52].
Engineering Strategy: From Plant to Microbe
The core concept of the semi-synthetic approach was to functionally transfer the artemisinic acid biosynthetic pathway from the plant A. annua into the industrially robust yeast Saccharomyces cerevisiae [52]. This involved several key engineering steps:
- Enhancing Precursor Supply: Engineering the native yeast MVA pathway to dramatically increase the flux of cytosolic acetyl-CoA toward the universal terpenoid precursor, FPP.
- Introducing Heterologous Pathway Enzymes: Integrating plant-derived genes (e.g., ADS, CYP71AV1, CPR) into yeast to redirect FPP from sterols toward amorpha-4,11-diene and its oxidation to artemisinic acid.
- Optimizing the Host Chassis: Fine-tuning the expression of pathway enzymes, their stoichiometry, and co-factors to maximize flux and minimize the accumulation of intermediates or toxic by-products.
Semi-Synthetic Production Methodology
The successful semi-synthetic production of artemisinin can be broken down into two major experimental components: the microbial production of artemisinic acid and its subsequent chemical conversion to artemisinin.
Microbial Production of Artemisinic Acid
Strain Engineering and Fermentation
The engineering of S. cerevisiae focused on rewriting its native metabolic network to overproduce artemisinic acid.
Protocol: Engineering High Flux to FPP
- Background: Wild-type yeast tightly regulates the MVA pathway for sterol biosynthesis.
- Method: Overexpress the enzymes of the MVA pathway, notably tHMG1 (a truncated, stabilized version of HMG-CoA reductase), to deregulate and enhance carbon flux from acetyl-CoA to FPP [52] [47].
- Result: This foundational step created a base strain capable of producing high titers of FPP.
Protocol: Expressing the Artemisinin-Specific Pathway
- Amorphadiene Production: Introduce the plant gene AaADS (Amorpha-4,11-diene Synthase) into the high-FPP strain. ADS diverts FPP from sterol production to amorpha-4,11-diene [52]. Initial titers of ~100 mg/L were improved to over 40 g/L in fed-batch fermentations through further strain optimization [52].
- Oxidation to Artemisinic Acid:
- Introduce the three-component oxidation system from A. annua: CYP71AV1 (P450), its cognate reductase AaCPR, and cytochrome b5 [52].
- Co-express the downstream enzymes ADH1 and ALDH1 to facilitate the efficient conversion of artemisinic alcohol and aldehyde intermediates to artemisinic acid [52].
- A critical optimization was fine-tuning the expression ratio of CYP71AV1 to AaCPR to improve electron transfer efficiency and reduce the formation of reactive oxygen species [52].
- Fermentation Process: The engineered strain was cultivated in a fed-batch bioreactor. To manage the high metabolic load and oxygen demand, a feed strategy utilizing a carbon source like ethanol or a glucose/ethanol mix was employed [52]. In situ product removal using oils like isopropyl myristate was also used to boost production by reducing potential feedback inhibition or cytotoxicity [52].
Quantitative Performance of Engineered Strains
Table 2: Production Metrics for Artemisinin Precursors in Engineered Yeast.
| Strain / System | Key Genetic Modifications | Product | Titer | Scale | Citation Context |
|---|---|---|---|---|---|
| Early Engineered Yeast | MVA pathway upregulation + ADS | Amorphadiene | ~100 mg/L | Lab-scale | [52] |
| Optimized Yeast | Enhanced MVA flux + ADS | Amorphadiene | > 40 g/L | Fed-batch (2L) | [52] |
| Commercial Production Strain | Full pathway (ADS, CYP71AV1, CPR, ADH1, ALDH1) + Cytochrome b5 | Artemisinic Acid | 25 g/L | Fed-batch (2L) | [52] |
Chemical Conversion to Artemisinin
The process for converting microbially produced artemisinic acid to artemisinin involves a multi-step chemical synthesis.
- Protocol: Chemical Synthesis from Artemisinic Acid [54]
- Reduction to Dihydroartemisinic Acid: Artemisinic acid is first hydrogenated to its saturated form, dihydroartemisinic acid (DHAA), which is the direct biosynthetic precursor to artemisinin.
- Photo-oxidation: The DHAA is then dissolved in an organic solvent (e.g., dichloromethane) and subjected to photo-oxidation. This step typically involves irradiating the solution with light in the presence of oxygen, a photosensitizer (e.g., tetraphenylporphyrin), and an acid catalyst (e.g., trifluoroacetic acid) [54].
- Mechanism: The photo-oxidation reaction cleaves oxygen molecules, generating singlet oxygen that attacks the DHAA backbone, leading to the formation of the characteristic endoperoxide bridge and subsequent rearrangement into artemisinin [54].
- Purification: The resulting artemisinin is then isolated and purified from the reaction mixture using standard techniques like chromatography and crystallization.
This chemical process was developed into a scalable industrial method, enabling the commercial production of semi-synthetic artemisinin which began in 2013 [52].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Semi-Synthetic Artemisinin R&D.
| Reagent / Material | Function / Application | Technical Context |
|---|---|---|
| Engineered S. cerevisiae Strain | Microbial chassis for artemisinic acid production. | Strain engineered with optimized MVA pathway, ADS, CYP71AV1, CPR, ADH1, ALDH1, and cytochrome b5 [52]. |
| pEAQ-based Vectors | Plant expression vectors for pathway gene characterization. | Used in A. annua for transient overexpression or silencing of artemisinin biosynthetic genes [51]. |
| AaADS, AaCYP71AV1, AaCPR Genes | Core pathway enzymes for heterologous expression. | ADS cyclizes FPP; CYP71AV1/CPR oxidize amorphadiene [52] [47]. Codon-optimization for yeast is critical. |
| Isopropyl Myristate | In situ product removal agent. | An oil overlay used in fermentations to sequester lipophilic artemisinic acid, reducing feedback inhibition and cytotoxicity [52]. |
| Tetraphenylporphyrin (TPP) | Photosensitizer in chemical synthesis. | Catalyzes the generation of singlet oxygen from triplet oxygen during the light-driven conversion of DHAA to artemisinin [54]. |
| Trifluoroacetic Acid (TFA) | Acid catalyst in chemical synthesis. | Used in the photo-oxidation reactor to promote the cyclization and rearrangement reactions forming artemisinin [54]. |
| Artemisinin ELISA Kit | Quantification of artemisinin in samples. | Immunoassay for rapid, high-throughput measurement of artemisinin and its derivatives, useful for quality control [55]. |
Impact and Future Perspectives
The successful development of semi-synthetic artemisinin represents a landmark achievement in synthetic biology and metabolic engineering. Its commercial production, initiated in 2013, provided a second, stable source for this critical antimalarial drug, helping to buffer against supply shortages and price volatility associated with agricultural production [52]. This project demonstrated the feasibility of engineering complex plant metabolic pathways in microbial hosts for large-scale industrial production.
Future directions in this field focus on further optimizing production and combating the emerging threat of artemisinin partial resistance in malaria parasites [50]. Key research areas include:
- Advanced Metabolic Engineering: Further optimization of yeast strains to utilize cheaper carbon sources (e.g., glucose directly) and achieve even higher titers of artemisinic acid, for instance by adapting strategies from high-producing β-farnesene strains [52].
- Addressing Resistance: The rise of PfK13 propeller domain mutations associated with delayed parasite clearance in Southeast Asia and Africa underscores the need for vigilant monitoring and new therapeutic strategies [50]. Research into non-artemisinin-based therapies and triple ACTs (TACTs) is ongoing [53] [49].
- Synergistic Approaches: Recent studies highlight the role of flavonoids in A. annua, which can synergize with artemisinin to enhance antimalarial efficacy and potentially delay resistance [56]. Future metabolic engineering may focus on co-producing artemisinin and specific flavonoids.
In conclusion, the semi-synthetic artemisinin project is a paradigm for how harnessing biosynthetic building blocks from primary metabolism in an engineered host can solve a critical global health challenge. It provides a robust framework for the production of other complex plant-derived natural products.
Overcoming Production Hurdles: Troubleshooting and Optimizing Biosynthetic Pathways
Addressing Metabolic Bottlenecks and Low Titer Yields in Engineered Systems
Achieving high titers, yields, and productivity (TYP) in engineered biological systems remains a fundamental challenge in industrial biotechnology. Metabolic bottlenecks—rate-limiting steps in biosynthetic pathways—frequently impede carbon flux toward desired products, resulting in suboptimal production efficiency and compromised economic viability. These bottlenecks often arise from inherent regulatory mechanisms, imbalanced enzyme expression, cofactor limitations, and substrate toxicity that collectively constrain metabolic flux. Within the context of biosynthetic building blocks derived from primary metabolism, these limitations become particularly pronounced in complex pathways such as the shikimate and aromatic amino acid biosynthesis routes, which serve as foundational platforms for numerous high-value compounds [57] [58]. Addressing these constraints requires systematic approaches that combine advanced genetic tools, computational modeling, and high-throughput screening technologies to identify and overcome critical pathway limitations.
The economic implications of unresolved metabolic bottlenecks are substantial, particularly for natural products with pharmaceutical relevance and renewable chemicals derived from lignocellulosic biomass. As production scales increase, even minor inefficiencies in pathway flux can significantly impact manufacturing costs and sustainability metrics [59]. This technical guide examines the core principles and methodologies for diagnosing, understanding, and resolving metabolic bottlenecks, with particular emphasis on applications within primary metabolic pathways that generate essential biosynthetic building blocks.
Systematic Identification of Metabolic Bottlenecks
Analytical Approaches for Bottleneck Detection
Accurately identifying metabolic bottlenecks requires multi-faceted analytical approaches that interrogate pathway functionality at molecular, enzymatic, and flux levels. Metabolite profiling through LC-MS or GC-MS provides direct evidence of pathway intermediates that accumulate at nodes of constrained flux, while transcriptomic and proteomic analyses reveal discrepancies between gene expression, protein abundance, and actual metabolic throughput [57]. For instance, in tyrosine biosynthesis studies with CHO cells, researchers identified critical bottlenecks by correlating intracellular tyrosine pools with transcriptional levels of key pathway enzymes including phenylalanine hydroxylase (PAH) and pterin-4α-carbinolamine dehydratase (PCBD1) [57].
Metabolic flux analysis (MFA) represents another powerful methodology for quantifying carbon channeling through different pathway branches and identifying nodes with limited capacity. By employing 13C-labeling techniques and computational modeling, MFA enables researchers to map absolute metabolic fluxes and pinpoint enzymatic steps that constrain overall pathway efficiency. In shikimate pathway engineering, this approach has revealed significant flux limitations at the 3-dehydroquinate synthase (aroB) and 3-dehydroquinate dehydratase (aroQ) steps, guiding subsequent optimization efforts [58].
Table 1: Analytical Methods for Bottleneck Identification
| Method | Key Measured Parameters | Information Provided | Typical Workflow |
|---|---|---|---|
| Metabolite Profiling | Intermediate concentrations, Byproduct accumulation | Direct evidence of flux constraints, Thermodynamic limitations | Quenching → Extraction → LC-MS/GC-MS → Data analysis |
| Transcriptomics/Proteomics | mRNA expression levels, Protein abundance | Capacity constraints, Regulatory bottlenecks | RNA extraction/Protein isolation → Sequencing/MS → Correlation with flux data |
| Metabolic Flux Analysis | In vivo reaction rates, Pathway flux distribution | Quantitative flux maps, Identification of rate-limiting steps | 13C-labeling → Isotopomer analysis → Computational modeling → Flux calculation |
| Enzyme Activity Assays | Catalytic rates, Kinetic parameters | Intrinsic enzyme capacity, Cofactor limitations | Cell lysis → Substrate supplementation → Product measurement → Kinetic analysis |
Computational Prediction of Pathway Limitations
Advanced computational tools now enable a priori prediction of potential metabolic bottlenecks before extensive experimental work. Retrobiosynthesis platforms such as BioNavi-NP employ deep learning algorithms to predict biosynthetic pathways and identify potentially problematic enzymatic transformations [60]. These systems use transformer neural networks trained on both general organic and biosynthetic reactions to generate candidate biosynthetic routes from target molecules to simple building blocks, achieving top-10 prediction accuracy of 60.6% for single-step biosynthetic reactions [60].
The Biosynfoni molecular fingerprinting system represents another computational approach specifically designed for natural product research, using 39 biosynthetically relevant structural features to analyze chemical space and pathway relationships [38]. By capturing biosynthetic building blocks like amino acids and isoprene units, this method enables more accurate prediction of biosynthetic distances between compounds, allowing researchers to identify pathway segments that may present engineering challenges [38].
Engineering Strategies to Overcome Metabolic Bottlenecks
Pathway Optimization and Enzyme Engineering
Combinatorial pathway optimization represents a powerful strategy for addressing metabolic bottlenecks without requiring complete mechanistic understanding of pathway limitations. The Statistical Design of Experiments (DoE) framework enables efficient exploration of complex gene expression landscapes with minimal experimental iterations [58]. In a case study optimizing para-aminobenzoic acid (pABA) production in Pseudomonas putida, researchers applied a Plackett-Burman design to modulate expression levels of all nine genes in the shikimate and pABA biosynthesis pathways, testing only 16 strain variants from a theoretical library of 512 combinations [58].
This systematic approach identified 3-dehydroquinate synthase (aroB) as a critical bottleneck in pABA biosynthesis, enabling targeted optimization that increased titers from initial screening values (2-186.2 mg/L) to a maximum of 232.1 mg/L through a second round of strain engineering [58]. The methodology employed characterized biological parts—promoters, ribosome binding sites (RBS), and plasmid origins of replication—with defined expression strengths to create predictable expression variants [58].
Table 2: Key Research Reagent Solutions for Metabolic Engineering
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Expression Modulators | JE111111 promoter (strong), JE151111 promoter (moderate), JER04 RBS (strong), JER10 RBS (moderate) | Fine-tuning gene expression levels in metabolic pathways |
| Vector Systems | pSEVA231 (medium copy, ~30), pSEVA621 (low copy, ~20) | Controlling gene dosage in pathway engineering |
| Computational Tools | BioNavi-NP, Biosynfoni fingerprint | Predicting biosynthetic pathways and analyzing natural product chemical space |
| Biosensors | Transcription factor-based biosensors (TetR, TrpR), Whole-cell biosensors | Real-time metabolite monitoring and high-throughput screening |
| Pathway Enzymes | Phenylalanine hydroxylase (PAH), Pterin-4α-carbinolamine dehydratase (PCBD1), 3-dehydroquinate synthase (aroB) | Key catalytic functions in targeted metabolic pathways |
Cofactor Engineering and Cofactor-Coupled Metabolite Balancing
Cofactor limitations frequently create hidden metabolic bottlenecks that are not apparent from simple pathway analysis. In tyrosine biosynthesis engineering, the essential tetrahydrobiopterin (BH4) regeneration cycle—mediated by PCBD1 and quinoid dihydropteridine reductase (QDPR)—proved critical for sustaining phenylalanine hydroxylase (PAH) activity and enabling endogenous tyrosine production [57]. This cofactor-coupled system requires balanced expression of multiple enzyme components to maintain functional flux.
Similarly, in lignocellulosic conversion systems, redox cofactor imbalances often constrain efficient substrate utilization. Engineering NADPH regeneration systems or implementing transhydrogenase cycles can alleviate such limitations and enhance pathway performance [59]. The integration of cofactor engineering with traditional pathway optimization represents a holistic approach to addressing interconnected metabolic constraints.
Dynamic Regulation and Biosensor-Enabled Control
Biosensors provide powerful tools for implementing dynamic metabolic control that automatically responds to pathway intermediate accumulation—a direct manifestation of metabolic bottlenecks. These systems typically employ transcription factor-based regulators that detect specific metabolites and modulate expression of bottleneck enzymes accordingly [59]. For instance, biosensors responsive to aromatic amino acids or shikimate pathway intermediates can dynamically regulate carbon influx or enzyme expression to balance flux distribution.
The development process for effective biosensors involves sensing module optimization (promoter engineering, RBS modification, operator tuning) and output module specification (fluorescent reporters, enzyme cascades, growth selection markers) [59]. When integrated with high-throughput screening systems, biosensor-enabled approaches allow rapid identification of bottleneck-alleviating variants from combinatorial libraries, dramatically accelerating the strain optimization process.
Figure 1: Integrated Framework for Addressing Metabolic Bottlenecks. This workflow outlines a systematic approach from bottleneck identification through intervention strategies to performance enhancement.
Case Studies: Successful Bottleneck Resolution in Biosynthetic Pathways
Tyrosine Biosynthesis in CHO Cells
Chinese hamster ovary (CHO) cells represent the predominant host system for monoclonal antibody production, but their limited endogenous tyrosine biosynthesis capacity creates significant challenges in high-density cultures [57]. The low solubility of tyrosine in neutral media further complicates exogenous supplementation strategies. Researchers addressed this bottleneck through metabolic engineering of the complete tyrosine biosynthesis pathway, focusing on the BH4-dependent conversion of phenylalanine to tyrosine.
The engineering strategy involved overexpression of PAH and PCBD1 to enhance the core hydroxylation and cofactor regeneration steps [57]. Experimental protocols included:
- Serial passage cultures in tyrosine-free media to identify clones with restored autotrophic capacity
- Fed-batch bioreactor studies comparing growth and productivity across tyrosine concentrations (0-3.0 mM)
- qPCR and western blot analyses to quantify transcriptional and translational changes in pathway enzymes
Engineered clones demonstrated significantly improved performance in tyrosine-free cultures, with specific growth rates comparable to supplemented controls (0.64-0.77 dâ»Â¹) and maintained viability >90% [57]. This approach reduced dependence on exogenous tyrosine supplementation and mitigated the accumulation of inhibitory phenylalanine derivatives.
Shikimate Pathway Optimization for pABA Production
The shikimate pathway serves as a fundamental aromatic building block source, but its complex regulation and multiple branch points create numerous potential bottleneck nodes. In pABA production using Pseudomonas putida, researchers implemented a combinatorial engineering approach to systematically identify and overcome pathway limitations [58].
The experimental methodology encompassed:
- Library design using a Plackett-Burman matrix covering 9 pathway genes at two expression levels (high/low)
- Strain construction with characterized genetic parts (promoters, RBS, origins of replication) controlling each gene
- Regression modeling of production data to identify significant gene effects
- Iterative strain engineering based on model predictions
This systematic approach revealed aroB (3-dehydroquinate synthase) as the principal bottleneck, with secondary limitations at aroE (shikimate dehydrogenase) and pabB (pABA synthase) nodes [58]. Targeted optimization of these specific steps enabled a substantial titer improvement to 232.1 mg/L, demonstrating the power of systematic bottleneck identification and resolution.
Figure 2: Shikimate Pathway with pABA Branch Highlighting Key Bottleneck. The aroB enzyme (red) was identified as the primary flux constraint in pABA production [58].
Emerging Technologies and Future Perspectives
Integration of Machine Learning and Adaptive Laboratory Evolution
The convergence of biosensor technology, systems biology, and machine learning is driving the next generation of metabolic engineering strategies [59]. These integrated systems enable continuous, data-driven optimization of pathway performance through iterative design-build-test-learn cycles. For example, biosensor-enabled high-throughput screening can generate training datasets for machine learning models that predict optimal gene expression configurations for minimizing bottleneck effects.
Adaptive laboratory evolution coupled with biosensor-mediated selection pressure represents another promising approach for bottleneck resolution without requiring detailed pathway understanding. By applying selective pressure based on product formation or intermediate detoxification, microbial populations can evolve enhanced flux through constrained pathway segments via mutational mechanisms that might not be intuitively designed.
Advanced Fermentation Strategies for Bottleneck Mitigation
Beyond genetic interventions, innovative bioprocess engineering approaches can help overcome metabolic bottlenecks, particularly those related to nutrient limitations or byproduct inhibition. In recombinant protein production systems, uncoupling protein production from growth through controlled nutrient limitation has demonstrated potential for enhancing product yields [61].
Experimental protocols for growth-decoupled production include:
- Retentostat or perfusion cultivation for maintaining high cell densities at near-zero growth rates
- Promoter engineering to utilize stress-responsive expression systems (e.g., PHSP12) that activate under slow-growth conditions
- Dynamic nutrient feeding strategies that separately optimize growth and production phases
Studies in Saccharomyces cerevisiae have shown that promoter selection critically influences production performance under slow-growing conditions, with stress-induced promoters (PHSP12) enhancing intracellular protein titers by 10-fold at very low growth rates, while constitutive promoters (PTEF1) improved secretion efficiency [61]. These findings highlight the importance of matching genetic design with process optimization for comprehensive bottleneck resolution.
Addressing metabolic bottlenecks and low titer yields requires integrated approaches that combine systematic identification methods with targeted intervention strategies. The continuing development of advanced analytical techniques, computational prediction tools, and high-throughput engineering platforms is progressively enhancing our ability to diagnose and resolve flux constraints in engineered biological systems. As these technologies mature, their application to biosynthetic building block production from primary metabolism will play a crucial role in enabling sustainable manufacturing paradigms for pharmaceuticals, chemicals, and materials.
Strategies for Balancing Precursor Pool Allocation and Minimizing Metabolic Burden
A central challenge in constructing efficient microbial cell factories lies in the inherent conflict between overproducing a target compound and maintaining cellular viability. The host cell's metabolic network is a finely tuned system, and rewiring it for biosynthesis often disrupts the delicate balance of precursor pool allocation, imposing a significant metabolic burden. This burden manifests as reduced growth rates, decreased protein synthesis capacity, and overall impaired host fitness, ultimately limiting the yield and productivity of the desired product [62] [63]. Within the broader context of biosynthetic building blocks from primary metabolism, achieving optimal production requires sophisticated strategies that dynamically manage the allocation of central metabolites—such as acetyl-CoA, malonyl-CoA, and amino acids—toward heterologous pathways without compromising essential cellular functions. This guide details the core principles and methodologies for achieving this critical balance, enabling the development of robust, high-yielding microbial production systems for pharmaceuticals, chemicals, and fuels.
Computational Frameworks for Predictive Modeling
Constraint-Based Modeling and Resource Allocation
Constraint-based metabolic models, particularly Genome-Scale Metabolic Models (GEMs), are indispensable tools for predicting cellular behavior after genetic modifications. These models comprehensively represent an organism's metabolism by integrating all metabolic reactions annotated from its genome [62]. A key advancement in this area is the explicit incorporation of Resource-Allocation Constraints (RACs), which govern the structure and function of metabolic networks by accounting for the limited availability of cellular resources, such as enzymes and ribosomes [63].
RACs implement simple, mechanism-agnostic limitations on the total flux through metabolic pathways, reflecting the reality that the cell's machinery for protein synthesis is finite. Studies have demonstrated that models incorporating RACs are significantly better at predicting interspecies interactions in microbial communities and simulating realistic growth phenotypes, as they prevent the model from allocating impossible levels of resources to metabolic processes [63]. For the metabolic engineer, this means that RAC-enabled models can more reliably identify engineering targets that enhance product yield without collapsing central metabolism due to excessive burden.
Quantitative Heterologous Pathway Design
The Quantitative Heterologous Pathway Design algorithm (QHEPath) represents a specialized computational approach for evaluating and designing pathways that break the native stoichiometric yield limits of a host organism [62]. This method relies on a high-quality Cross-Species Metabolic Network model (CSMN), which is constructed by integrating biochemical reactions from multiple species into a unified framework. The CSMN model undergoes rigorous quality control to eliminate errors, such as the infinite generation of reducing equivalents or energy, which would otherwise lead to unrealistic yield predictions [62].
The QHEPath algorithm systematically calculates the potential yield improvement for a target product by introducing heterologous reactions. It distinguishes between the minimal reactions needed to make a non-native product and the additional reactions that specifically serve to exceed the host's native yield limit. A large-scale evaluation of 12,000 biosynthetic scenarios across 300 products revealed that over 70% of product pathway yields could be improved by introducing appropriate heterologous reactions, leading to the identification of 13 common engineering strategies categorized as carbon-conserving and energy-conserving [62]. This tool provides a quantitative framework for prioritizing pathway designs that efficiently utilize precursor pools.
Table 1: Key Computational Tools and Their Applications in Alleviating Metabolic Burden
| Tool/Algorithm | Core Function | Application in Balancing Precursors & Burden |
|---|---|---|
| Genome-Scale Metabolic Models (GEMs) with RACs [63] [64] | Simulates flux distributions in a metabolic network under physicochemical constraints. | Predicts how resource limitations affect growth and production; identifies gene knockout/knock-in targets that minimize burden. |
| Quantitative Heterologous Pathway Design (QHEPath) [62] | Calculates yield potential and identifies heterologous reactions to break native yield limits. | Pinpoints carbon- and energy-conserving pathways that enhance yield without disproportionately draining precursor pools. |
| Cross-Species Metabolic Network (CSMN) [62] | Integrated model containing a diverse array of biochemical reactions from multiple species. | Provides a validated reaction database for designing efficient heterologous pathways in non-native hosts. |
Key Engineering Strategies for Pathway Optimization
Precursor Overproduction and Channeling
A foundational strategy is to engineer the host to overproduce key precursor metabolites from primary metabolism. This involves enhancing the flux through central carbon pathways (e.g., glycolysis, pentose phosphate pathway) to ensure an abundant supply of building blocks like acetyl-CoA, phosphoenolpyruvate, and erythrose-4-phosphate [65]. The use of platform strains that already overproduce these central metabolites or key secondary metabolite intermediates (e.g., (S)-reticuline for alkaloids) can dramatically accelerate project timelines by providing a optimized starting point [65].
To further improve efficiency, metabolic channeling can be engineered. This concept involves co-localizing sequential enzymes in a pathway to facilitate the direct transfer of intermediates between active sites, minimizing diffusion losses, reducing intermediate toxicity, and protecting unstable intermediates from degradation. This approach effectively increases the local concentration of precursors for downstream enzymatic steps, thereby enhancing overall pathway flux.
Dynamic Pathway Regulation
Static overexpression of pathway genes often leads to metabolic imbalance and excessive burden. Precision metabolic engineering offers a solution through the design of systems that dynamically regulate pathway flux in response to cellular or environmental signals [66]. This involves three key hallmarks: sensing specific signals, completely directing metabolic flux based on those signals, and producing sharp responses at predetermined thresholds [66].
For example, pathways can be designed to remain inactive during the rapid growth phase, allowing the cell to build biomass without competition from the heterologous pathway. Once a sufficient cell density is reached, a sensory mechanism (e.g., a quorum-sensing circuit) can trigger the activation of the production pathway [66]. This dynamic control ensures that resource-intensive production occurs only when the cellular resource pool is sufficient, thereby minimizing the burden on growth.
Modular Pathway Optimization and Co-culture Systems
Complex pathways, especially for plant natural products (PNPs), can be broken down into smaller, optimized modules [65]. This "divide and conquer" strategy allows for the independent tuning of different pathway sections—such as the upstream precursor-forming module and the downstream derivatization module—before reintegrating them into a single host [65].
When pathway complexity or burden is too high for a single host, co-culture systems present a powerful alternative. Here, the total metabolic burden is distributed across multiple engineered microbial strains, each specialized in a specific part of the biosynthetic route [65]. For instance, in one study, the biosynthesis of benzylisoquinoline alkaloids (BIAs) was split between E. coli and S. cerevisiae, with each host performing the steps it was best suited for [65]. Success in co-cultures requires careful balancing of strain growth and efficient transport of pathway intermediates between the different organisms.
Experimental Protocols for Implementation
Protocol for Model-Driven Host Engineering
This protocol utilizes computational predictions to guide targeted genetic modifications.
- Model Reconstruction and Curation: Begin with an existing high-quality GEM for your host organism (e.g., E. coli or S. cerevisiae). For non-native products, employ an integrated model like CSMN to ensure a comprehensive reaction database [62].
- Simulation with Resource-Allocation Constraints (RACs): Implement RACs in your model to simulate realistic cellular limitations [63]. Set the biomass formation reaction as the objective function to simulate growth. Then, add a reaction for your target product and perform simulations (e.g., Flux Balance Analysis) to predict maximum theoretical yield.
- Identification of Engineering Targets: Use algorithms like OptKnock to identify gene knockout candidates that couple growth with product formation. Simultaneously, use tools like QHEPath to find heterologous reactions that conserve carbon or energy and improve yield beyond the native limit [62].
- Genetic Implementation: In the lab, execute the predicted gene knockouts using techniques such as CRISPR-Cas9 or lambda Red recombinase. Introduce heterologous genes via plasmids or genomic integration.
- Validation and Iteration: Measure the product titer, yield, and productivity of the engineered strain in a controlled bioreactor. Quantify metabolic burden through growth rate and biomass yield measurements. Use these experimental data to refine and re-parametrize the metabolic model for the next cycle of the Design-Build-Test-Learn (DBTL) loop [64].
Protocol for Dynamic Pathway Control Implementation
This protocol outlines the steps for building a dynamically regulated pathway.
- Sensor Selection: Choose a sensory protein that responds to a desired intracellular or extracellular signal. This could be a transcription factor that senses a quorum-sensing molecule, a specific metabolite (e.g., acetyl-CoA), or an exogenous inducer [66].
- Promoter Engineering: Place the key, rate-limiting genes of your heterologous pathway under the control of a promoter that is regulated by the chosen sensor. This creates a genetic circuit where pathway expression is tied to the signal.
- Circuit Characterization: Transform the genetic construct into your host and characterize the input-output relationship. Measure product formation and growth rate at different signal concentrations to determine the activation threshold and dynamic range [66].
- Fine-Tuning the Response: If the response is not sharp enough, employ strategies to improve sensitivity. This can include tuning the copy number of the sensor gene, incorporating hybrid promoters, or adding regulatory cascades to amplify the signal [66].
- Bioreactor Cultivation: Cultivate the engineered strain in a bioreactor. For an autonomous system, allow the culture to grow until the triggering metabolite or population density is reached. For a user-controlled system, add the chemical inducer at the optimal time. Monitor the timing and level of product synthesis relative to growth.
Visualization of Core Workflows and Strategies
Metabolic Engineering Workflow Diagram
Diagram 1: The iterative Design-Build-Test-Learn (DBTL) cycle in metabolic engineering, driven by computational models and experimental validation.
Strategies for Balancing Precursors and Burden
Diagram 2: Key engineering strategies for balancing precursor supply and minimizing metabolic burden.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 2: Key Reagents and Materials for Metabolic Engineering Experiments
| Reagent/Material | Function/Application | Example Use-Case |
|---|---|---|
| Platform Strains [65] | Engineered hosts that overproduce central precursors (e.g., acetyl-CoA, malonyl-CoA, (S)-reticuline). | Provides a high-flux starting point for pathways utilizing a specific precursor, saving extensive engineering time. |
| Cross-Species Metabolic Model (CSMN) [62] | A quality-controlled, integrated metabolic database. | Used to design heterologous pathways and identify carbon/energy-conserving reactions that break yield limits. |
| Resource-Allocation Constraint (RAC) Models [63] | Genome-scale models incorporating enzyme and ribosome limitations. | Predicts realistic flux distributions and growth phenotypes, preventing designs that overburden the host. |
| Inducible Promoter Systems & Genetic Sensors [66] | Enable dynamic control of gene expression in response to chemical or metabolic signals. | Used to decouple growth and production phases, expressing pathways only when cellular resources are abundant. |
| CRISPR-Cas9 Tools | For precise gene knockouts, knock-ins, and multiplexed genome editing. | Essential for implementing model-predicted gene deletions and integrating heterologous pathways into the host genome. |
| Analytical Standards & LC-MS/MS | For quantifying target products, intermediates, and intracellular metabolites. | Critical for validating model predictions, calculating yields, and identifying metabolic bottlenecks or imbalances. |
The successful engineering of microbial cell factories hinges on a sophisticated understanding of the intrinsic trade-offs between precursor pool allocation and metabolic burden. By leveraging predictive computational models that account for resource constraints, implementing dynamic control systems, and employing modular design principles, researchers can create robust production strains. The integration of these strategies within an iterative DBTL framework, supported by the toolkit of reagents and analytical methods, provides a systematic path forward. This approach is fundamental to advancing the production of biosynthetic building blocks from primary metabolism, ultimately enabling the efficient and scalable synthesis of high-value pharmaceuticals and chemicals.
Overcoming Enzyme Specificity and Cross-Talk in Complex Pathway Engineering
The engineering of complex biosynthetic pathways represents a frontier in synthetic biology, enabling the production of high-value chemicals, pharmaceuticals, and biofuels. However, two fundamental biological phenomena consistently challenge these efforts: inherent enzyme specificity and pervasive pathway cross-talk. Enzyme specificity, while crucial for metabolic fidelity in native systems, often limits the ability of engineered pathways to utilize non-native substrates. Simultaneously, cross-talk—the unintended interaction between engineered and endogenous cellular networks—can divert metabolic flux, create toxic intermediates, and destabilize entire pathways. Within the context of biosynthetic building blocks derived from primary metabolism, these challenges become particularly pronounced. Primary metabolism provides essential precursor pools, such as malonyl-CoA, acetyl-CoA, and various amino acids, which are the foundation for both essential cellular functions and engineered pathways for secondary metabolites [67]. This technical guide examines the underlying mechanisms of these challenges and presents a suite of advanced computational, experimental, and systems-level strategies to overcome them, thereby enabling the robust engineering of complex metabolic networks.
Defining the Problem Space: Specificity and Cross-Talk Mechanisms
Molecular Foundations of Enzyme Specificity
Enzyme specificity is governed by the precise molecular architecture of the active site. The classic Lock and Key Model, proposed by Emil Fischer, posits that the enzyme's active site is a rigid structure complementary in shape and chemical properties to its substrate [68]. This model explains several specificity levels:
- Absolute Specificity: The enzyme catalyzes a reaction for only one substrate.
- Group Specificity: The enzyme acts on molecules with specific functional groups.
- Stereochemical Specificity: The enzyme distinguishes between different stereoisomers.
A more dynamic perspective is offered by the Induced Fit Model, where the active site undergoes conformational changes upon substrate binding to form a complementary fit [68]. This flexibility allows some enzymes to accommodate multiple substrates but also creates potential for off-target activity in engineered contexts.
Metabolic Cross-Talk: Forms and Consequences
Cross-talk in metabolic engineering manifests in several forms, each with distinct consequences:
- Precursor Competition: Engineered pathways compete with endogenous metabolism for shared building blocks. For instance, polyketide synthases (PKS) and fatty acid synthases (FAS) both utilize malonyl-CoA as an essential extender unit, creating direct competition for this central metabolite [67].
- Regulatory Interference: Engineered enzymes may be inadvertently regulated by endogenous metabolites. Research in Saccharomyces cerevisiae has revealed extensive regulatory cross-talk where metabolites from disparate pathways activate enzymes, forming a network-wide regulatory system [69].
- Inhibitory Interactions: Endogenous metabolites may inhibit heterologous enzymes. Alarmingly, one study found that up to 54% of enzymatic reactions in yeast could be intracellularly activated by metabolites, suggesting an equally extensive potential for inhibition [69].
- Electron Transfer Competition: Pathways relying on redox reactions often compete for shared pools of electron carriers (NAD(P)H, FADH2), creating imbalanced cofactor regeneration.
Table 1: Quantitative Analysis of Enzyme-Metabolite Interactions in S. cerevisiae
| Interaction Type | Percentage of Enzymes | Number of Metabolites Involved | Key Characteristics |
|---|---|---|---|
| Intracellular Activation | 54% (344/635) | 286 | Forms extensive trans-activation network between pathways |
| Extracellular Molecule Activation | 19% (121/635) | Not specified | Potential for non-native inducers |
| No Known Activation | 27% (170/635) | N/A | Potential targets for novel engineering |
| Lipids as Activators | Low prevalence | Low | High prevalence in inhibitory interactions |
Computational Strategies for Predicting and Engineering Specificity
Integrative Genomic Mining for Novel Enzyme Function
The discovery of enzymes with desired specificities from natural sequence space can be dramatically accelerated through computational mining. A pioneering approach, Integrative Genomic Mining, successfully identified ketoacid decarboxylases specific for long-chain (C5-C8) substrates from a family of over 17,000 sequences [70]. The methodology involves a multi-stage filtration process:
- Sequence Homology Search: Initial identification of genomic enzyme orthologues (GEOs) based on sequence similarity to a query enzyme of interest.
- Redundancy and Host Compatibility Filtering: Removal of redundant sequences (>90% identity) and sequences from incompatible hosts (e.g., eukaryotes for bacterial expression).
- Structural Homology Modeling: Generation of 100+ homology models for each GEO using tools like Rosetta Comparative Modeling, followed by selection of the lowest-energy model.
- Active Site Docking and Design: Docking of the desired substrate (e.g., C8 ketoacid) into the predicted active site, allowing for conservative mutations to optimize the protein-ligand interface energy.
This pipeline enriched for active enzymes, yielding a set where the median catalytic efficiency was 75-fold greater than naively selected homologues. The top-performing enzyme, GEO 175, exhibited a 33,000-fold higher catalytic efficiency for C8 over C3 substrates [70].
Biosynfoni: A Biosynthesis-Informed Fingerprint for Pathway Design
Predicting the behavior of novel enzymatic pathways requires understanding biosynthetic relatedness. The Biosynfoni molecular fingerprint addresses this by explicitly encoding biosynthetic building blocks—such as common amino acids and isoprene units—into a compact, 39-substructure key array [38]. Unlike traditional fingerprints, Biosynfoni more accurately captures biosynthetic distance (the number of enzymatic steps separating two compounds), with similarity scores continuously decreasing as the number of reaction steps between compound pairs increases. This allows researchers to:
- Visualize Chemical Space: Map natural products and pathway intermediates based on biosynthetic logic.
- Predict Pathway Compatibility: Identify potential cross-talk by assessing the biosynthetic proximity of engineered pathways to native metabolic networks.
- Interpret Machine Learning Models: The concrete, biologically relevant substructure keys make classification decisions in models interpretable, crucial for diagnosing engineering problems [38].
Integrative Genomic Mining Workflow
Experimental Methodologies for Characterizing and Mitigating Cross-Talk
Protocol: Mapping the Enzyme Activation Network
To systematically identify potential regulatory cross-talk before pathway engineering, the following protocol, adapted from a genome-scale study in yeast, can be employed [69]:
Network Reconstruction:
- Input: A genome-scale metabolic model (e.g., Yeast9 for S. cerevisiae).
- Data Integration: Map cross-species enzyme kinetic data from the BRENDA database onto the model. For each enzyme, download all associated activator molecules.
- Curation: Remove non-cellular molecules (drugs, assay compounds) by comparing the activator list with the model's known intracellular metabolites.
- Output: A cell-intrinsic activation network where nodes are enzymes and metabolites, and edges represent activation relationships.
Network Analysis:
- Identify "hotspot" metabolites that activate multiple enzymes across different pathways, indicating high potential for cross-talk.
- Calculate pathway distances between the producing enzyme of an activator and the enzymes it activates. Short distances suggest rapid, local regulation, while long distances indicate system-wide signaling.
- Correlate enzyme essentiality with activation levels. The study found that highly activated enzymes are often non-essential, whereas the activating metabolites themselves are frequently essential [69].
Experimental Validation:
- Construct knockout strains for non-essential, highly activated enzymes.
- Measure metabolite levels (e.g., via LC-MS) and pathway fluxes (e.g., via 13C tracing) in knockout vs. wild-type strains under different nutrient conditions.
- The original study analyzed transcriptome profiles from >600 conditions and proteome profiles of hundreds of knockout strains to validate network predictions [69].
Protocol: Engineering Cross-Talk Compensation in Gene Circuits
When eliminating cross-talk at the molecular level is infeasible, a powerful alternative is to engineer compensatory circuits at the network level. This methodology was demonstrated in E. coli for reactive oxygen species (ROS) sensing [71].
Circuit Construction:
- Design two sensor circuits: one for the target signal (e.g., H2O2 via OxyR transcription factor) and one for the interfering signal (e.g., superoxide via SoxR transcription factor).
- Use medium- or high-copy plasmids to house the circuits, expressing the transcription factor constitutively and the fluorescent output (e.g., mCherry) under a cognate promoter (e.g., oxySp for OxyR).
- Assemble both circuits in a single host strain without modifying the chromosome.
Crosstalk Quantification:
- Expose the dual-sensor strain to both signals individually and in combination.
- Measure the output fluorescence for each sensor across a range of signal concentrations.
- Fit the dose-response curves to Hill functions and calculate the degree of crosstalk from the non-cognate signal.
Compensatory Circuit Design:
- Design a circuit that takes the output of the interfering-signal sensor and uses it to adjust the output of the target-signal sensor.
- This acts as an interference-cancellation circuit, mathematically subtracting the unintended contribution from the crosstalk.
- The final output reflects the concentration of the target signal with significantly reduced interference [71].
Cross-talk Compensation Principle
Case Studies in Primary and Secondary Metabolism Engineering
Reconciling Polyketide and Fatty Acid Biosynthesis
The interconnection between fatty acid synthase (FAS, primary metabolism) and polyketide synthase (PKS, secondary metabolism) is a classic example of inherent cross-talk. Both pathways:
- Share Precursors: Compete for malonyl-CoA and acetyl-CoA extender units.
- Use Similar Logic: Employ decarboxylative Claisen condensations and similar reductive processing domains (KR, DH, ER) [67].
- Co-regulate: Inhibition of the FAS enoyl reductase (FabI) by triclosan was found to also regulate the production of the polyketide actinorhodin in Streptomyces coelicolor [67].
Engineering Solutions:
- Precursor Pool Balancing: Overexpress acetyl-CoA carboxylase (ACC) to enhance malonyl-CoA supply, alleviating competition.
- Pathway Insulation: Use orthogonal acyl carrier proteins (ACPs) and cognate partner enzymes for the PKS to minimize interaction with the native FAS.
- Biosensor-Mediated Regulation: Implement malonyl-CoA biosensors to dynamically regulate pathway expression and maintain precursor homeostasis [67] [72].
Functional Expression of Challenging Metallocluster Enzymes
Metallocluster enzymes (e.g., those with FeS, FeMo, or NiFe clusters) are essential for many pathways but notoriously difficult to express functionally in heterologous hosts due to specific maturation requirements. Nitrogenases, hydrogenases, and radical SAM enzymes often exhibit little to no activity in standard industrial hosts [73].
Engineering Protocols:
Identify and Express Maturation Pathways:
- For iron-sulfur (FeS) clusters, co-express the requisite maturation machinery (e.g., the isc or suf operons from a native host).
- For [NiFe]-hydrogenases, express the hypABCDEF genes responsible for Ni insertion.
Enhance Electron Transfer:
- Identify and co-express the specific electron transfer proteins (ferredoxins, flavodoxins) required for the metalloenzyme's catalytic cycle.
- Engineer electron supply pathways to avoid competition with native metabolism.
Address Oxygen Sensitivity:
- Express oxygen-sensitive metalloenzymes under anaerobic conditions or in microaerophilic hosts.
- Engineer host strains with reduced oxidative stress (e.g., knockout ROS-generating genes) [73].
Table 2: Key Research Reagents for Overcoming Specificity and Cross-Talk
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
| BRENDA Database | Comprehensive repository of enzyme kinetic data (Km, kcat, activators, inhibitors). | Mapping potential regulatory cross-talk during pathway design [69]. |
| Rosetta Modeling Suite | Software for comparative modeling, protein-ligand docking, and enzyme design. | Reprogramming substrate specificity of ketoacid decarboxylase [70]. |
| Biosynfoni Fingerprint | A biosynthesis-informed molecular fingerprint (39 substructure keys). | Predicting biosynthetic distance and potential pathway interference [38]. |
| CRISPR-Cas9 | Tool for precise genomic modifications and multiplexed gene knockouts. | Knocking out endogenous genes to insulate heterologous pathways from cross-talk [72]. |
| FeS Cluster Maturation Systems (ISC, SUF) | Operons for assembling and inserting iron-sulfur clusters into apoenzymes. | Enabling functional expression of heterologous metalloenzyme pathways [73]. |
| Biosensors (e.g., malonyl-CoA) | Genetic circuits that report on or respond to metabolite concentration. | Dynamic regulation of pathway expression to maintain precursor balance [72]. |
Overcoming enzyme specificity and cross-talk is not merely a technical obstacle but a fundamental requirement for the reliable scale-up of complex pathway engineering. The strategies outlined—from integrative genomic mining and biosynthesis-informed design to network-level compensation circuits and precise host engineering—provide a comprehensive toolkit for addressing these challenges. The field is rapidly evolving, with several emerging trends poised to further advance capabilities:
- AI-Integrated Protein Design: Machine learning models, trained on vast sequence-structure-function datasets, are accelerating the de novo design of enzymes with bespoke specificities and minimal cross-reactivity [72] [4].
- Dynamic Metabolon Engineering: The spatial organization of enzymes into synthetic metabolons can channel intermediates effectively, minimizing off-target interactions and enhancing pathway flux [4].
- Advanced Host Engineering: The development of "chassis" strains with streamlined metabolisms, reduced regulatory complexity, and orthogonal resource allocation systems will provide cleaner backgrounds for heterologous pathway expression.
By adopting a holistic view that considers the engineered pathway within the context of the entire host metabolic network, researchers can transform specificity and cross-talk from debilitating problems into manageable design parameters. This shift is crucial for harnessing the full potential of primary metabolic building blocks to produce the next generation of biosynthetic products.
The efficient microbial production of high-value chemicals, such as pharmaceuticals, biofuels, and specialty compounds, often requires the re-routing of central metabolic fluxes. Native cellular metabolism, however, is a complex and highly regulated network designed for growth and survival, not for the maximal synthesis of a target compound. Competing pathways consume precious precursors and co-factors, diverting flux away from the desired product and limiting yield and productivity. Downregulating these competing pathways is therefore a critical step in metabolic engineering. RNA interference (RNAi) and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technologies have emerged as two powerful and distinct tools for achieving this targeted downregulation. Within the broader context of biosynthetic building blocks research, the strategic selection and application of these technologies enable researchers to sculpt cellular metabolism, enhancing the flow of carbon and energy from primary metabolism into engineered, high-value biosynthetic pathways. This whitepaper provides an in-depth technical guide for researchers and scientists on leveraging RNAi and CRISPR to silence competing metabolic genes, complete with comparative analysis, detailed protocols, and visual workflows.
Technology Fundamentals: Mechanisms of Gene Silencing and Editing
RNA Interference (RNAi): Targeted mRNA Knockdown
RNAi is a naturally occurring, conserved gene-silencing mechanism that regulates gene expression at the post-transcriptional level by degrading target messenger RNA (mRNA) or blocking its translation [74] [75]. The process can be harnessed experimentally by introducing exogenous double-stranded RNA (dsRNA) or synthetic small interfering RNAs (siRNAs) into cells.
Mechanism: The core machinery involves the enzyme Dicer, which processes long dsRNA or precursor microRNAs (pre-miRNAs) into short ~21-nucleotide RNA fragments. These small RNAs are then loaded into the RNA-induced silencing complex (RISC). The antisense ("guide") strand within RISC binds to complementary mRNA sequences. Upon perfect complementarity, the Argonaute protein within RISC cleaves the target mRNA, leading to its degradation. With imperfect pairing, translation is physically blocked, leading to reversible gene knockdown [75] [76].
Key Features for Metabolic Engineering: RNAi generates a knockdown effect, which is typically transient and reversible. This allows for dose-responsive studies of gene silencing, which is invaluable for investigating essential genes whose complete knockout would be lethal to the production host [76].
CRISPR-Cas Systems: Permanent DNA Modification
The CRISPR-Cas system, derived from a prokaryotic adaptive immune system, enables precise, permanent modifications to the genome itself [74] [77]. The most common system, CRISPR-Cas9, consists of two key components:
Mechanism: A guide RNA (gRNA) directs the Cas9 nuclease to a specific DNA sequence complementary to the gRNA. Cas9 then creates a double-strand break (DSB) in the target DNA. The cell repairs this break primarily via the error-prone non-homologous end joining (NHEJ) pathway, which often results in small insertions or deletions (indels) that disrupt the gene's coding sequence, leading to a permanent knockout [75] [76].
Key Features for Metabolic Engineering: Beyond knockout generation, CRISPR technology has been expanded to include powerful knockdown tools. CRISPR interference (CRISPRi) uses a catalytically "dead" Cas9 (dCas9) that lacks nuclease activity. The dCas9, guided by a gRNA, binds to a target DNA sequence without cutting it, physically obstructing transcription and leading to robust gene repression [74]. This offers a reversible silencing alternative to permanent knockouts.
Comparative Analysis: Selecting the Appropriate Tool
The choice between RNAi and CRISPR depends on the experimental goals, the nature of the target gene, and the desired outcome. The table below provides a structured comparison to guide this decision.
Table 1: Strategic Comparison of RNAi and CRISPR for Downregulating Competing Pathways
| Feature | RNAi (Knockdown) | CRISPR (Knockout/CRISPRi) |
|---|---|---|
| Mechanism of Action | Post-transcriptional; degrades or blocks mRNA translation [75] | DNA-level; introduces indels for knockout (Cas9) or blocks transcription (dCas9 for CRISPRi) [75] [76] |
| Genetic Outcome | Transient, reversible knockdown [76] | Permanent knockout or reversible repression (CRISPRi) [76] |
| Efficacy | Incomplete knockdown; residual gene expression is common [76] | Complete and permanent gene disruption is achievable with CRISPR-Cas9 [76] |
| Specificity | High risk of sequence-dependent and independent off-target effects [75] [78] | Higher specificity; advanced gRNA design and modified Cas variants minimize off-targets [76] [78] |
| Ideal Use Case | Silencing essential genes in a titratable manner; rapid, transient validation studies [76] | Complete elimination of non-essential competing pathways; multiplexed silencing; stable strain engineering [74] |
| Throughput | Well-suited for high-throughput screening, but confounded by off-target effects [75] | Superior for high-throughput genetic screens due to higher specificity and consistency [78] |
| Experimental Workflow | Relatively fast and simple; direct introduction of siRNAs or shRNA-encoding plasmids [76] | More complex; requires delivery of Cas nuclease and gRNA, often via plasmids or ribonucleoproteins (RNPs) [75] |
Table 2: Quantitative Comparison of On-Target and Off-Target Effects from a Large-Scale Study [78]
| Technology | On-Target Efficacy | Prevalence of Seed-Based Off-Target Effects | Correlation Between Reagents Targeting Same Gene |
|---|---|---|---|
| RNAi (shRNAs) | Effective knockdown observed | Strong and pervasive; a major component of the expression signature | Low |
| CRISPR (sgRNAs) | Comparable to RNAi | Negligible systematic off-target activity | High |
Experimental Protocols for Pathway Downregulation
RNAi-Mediated Silencing Protocol
This protocol outlines the process for using vector-derived short hairpin RNAs (shRNAs) to downregulate a target gene in a microbial host.
shRNA Design and Cloning:
- Design: Identify a 19-22 nt target sequence within the mRNA of the competing pathway gene. Use established design tools (e.g., from vendor libraries) to ensure specificity and minimize off-target effects by checking for homology to other genes.
- Cloning: Synthesize oligonucleotides encoding the shRNA sequence and clone them into an appropriate expression plasmid under a regulated promoter (e.g., inducible or constitutive).
Delivery: Transform the shRNA-encoding plasmid into your production host (e.g., E. coli or yeast) using standard methods like heat shock or electroporation.
Cultivation and Induction:
- Grow transformed cells in selective media.
- If using an inducible promoter, add the inducer (e.g., IPTG) to initiate shRNA expression.
- Continue cultivation to allow for gene silencing to occur.
Validation and Analysis:
- Efficiency Validation: Quantify knockdown efficiency using qRT-PCR to measure residual target mRNA levels and/or immunoblotting to assess protein levels.
- Phenotypic Screening: Measure the impact on the target biosynthetic pathway. This can include quantifying the reduced output of the competing metabolite and the increased production of the desired end-product [79]. For instance, in a pathway optimized for naringenin production, successful downregulation of a competing pathway would be confirmed via HPLC or LC-MS showing reduced byproduct accumulation and increased naringenin titer [79].
CRISPR-Cas9 Mediated Knockout Protocol
This protocol describes the use of CRISPR-Cas9 to create permanent knockouts of genes in a competing pathway.
gRNA Design and Vector Construction:
- Design: Design a gRNA sequence (typically 20 nt) targeting an early exon of the gene in the competing pathway to maximize the chance of a frameshift mutation. Use computational tools (e.g., CRISPR design software from Broad Institute) to predict on-target efficiency and minimize off-target sites.
- Construction: Clone the gRNA sequence into a plasmid that also expresses the Cas9 nuclease, often as a two-vector system or a single all-in-one vector.
Delivery: Co-transform or sequentially transform the Cas9 and gRNA plasmids into the production host.
Screening and Isolation:
- After transformation, plate cells on selective media. The DSB repair via NHEJ will generate a heterogeneous population of cells with various indels.
- Screen individual colonies for successful gene knockout. This can be done via:
- PCR and TIDE Analysis: Amplify the target genomic region and sequence the products. The TIDE (Tracking of Indels by DEcomposition) software can deconvolute the mixture of sequences and quantify the editing efficiency [80].
- Phenotypic Screening: Couple CRISPR with a biosensor selector. Engineer a sensor that links the concentration of a target chemical (the product of your biosynthetic pathway) to cell fitness (e.g., antibiotic resistance) [79]. Under selective pressure, only cells with improved production—potentially through successful knockout of a competing gene—will survive, allowing for direct evolution of optimized strains [79].
Validation: Confirm the knockout by Sanger sequencing of the target locus from isolated clones and verify the loss of protein function through enzymatic assays or metabolomic profiling.
Integrated Workflows and Visual Guide
The following diagrams illustrate the core mechanisms and a strategic experimental workflow for implementing these technologies.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for RNAi and CRISPR Experiments
| Reagent / Tool | Function | Example & Notes |
|---|---|---|
| siRNA / shRNA | The effector molecule that triggers sequence-specific mRNA degradation. | Synthetic siRNAs: For transient transfection. shRNA-encoding plasmids: For stable, long-term expression. |
| CRISPR-Cas9 System | The effector complex for DNA targeting. | All-in-one plasmids: Express both gRNA and Cas9. RNP complexes: Pre-assembled gRNA and Cas9 protein for high efficiency and reduced off-target effects [75]. |
| gRNA Design Tools | Computational software to predict efficient and specific guide RNAs. | Tools from the Broad Institute or commercial vendors help minimize off-target effects [75]. |
| Biosensor Selectors | Links intracellular metabolite concentration to cell survival for high-throughput screening. | An antibiotic resistance gene under the control of a metabolite-responsive promoter enriches for high-producing mutants during evolution experiments [79]. |
| Metabolite Analysis | Quantifies the success of pathway optimization by measuring product and byproduct levels. | LC-MS/MS or HPLC: For precise identification and quantification of target chemicals and pathway intermediates [28]. |
| NGS Analysis Tools | Validates editing efficiency and detects off-target effects. | Whole-genome sequencing and specialized algorithms (e.g., TIDE analysis for CRISPR) are critical for confirmation [80] [78]. |
The strategic downregulation of competing metabolic pathways is a cornerstone of modern metabolic engineering for the production of biosynthetic building blocks. Both RNAi and CRISPR offer powerful, yet distinct, solutions to this challenge. RNAi provides a reversible and titratable means to study essential gene functions and perform initial validation, while CRISPR technologies, including knockout and CRISPRi, offer permanent, highly specific, and multiplexable options for stable strain development. The integration of these tools with advanced screening methods, such as biosensor-coupled evolution, creates a robust framework for systematically optimizing microbial cell factories. By understanding the strengths and applications of each technology, as outlined in this guide, researchers can make informed decisions to accelerate the development of efficient and sustainable bioproduction platforms.
Proving and Improving: Validating Pathways and Comparing Biosynthetic Strategies
Tracking metabolic flux—the dynamic flow of metabolites through biochemical pathways—is fundamental to understanding how organisms convert primary metabolic building blocks into complex molecules. In biosynthetic building blocks research, this involves elucidating how simple precursors from central carbon and nitrogen metabolism are directed toward the synthesis of amino acids, nucleotides, lipids, and specialized secondary metabolites. The integration of metabolomics and transcriptomics has emerged as a powerful methodological framework for analytical validation in metabolic flux studies, enabling researchers to move beyond static snapshots to dynamic assessments of pathway activity [81] [82]. This approach provides systems-level validation of how genetic regulation translates into metabolic phenotype through enzyme activities and pathway fluxes.
This technical guide examines current methodologies, experimental designs, and analytical frameworks for employing multi-omics approaches to track metabolic flux, with particular emphasis on applications in primary metabolism and biosynthesis research. We focus specifically on practical implementation for researchers investigating metabolic pathway dynamics in both model and non-model organisms.
Conceptual Framework: Linking Multi-Omics Data to Metabolic Flux
Theoretical Foundations
Metabolic flux represents the integrated output of gene expression, protein activity, and metabolic regulation. Transcriptomics provides insights into potential metabolic capacity through expression of pathway enzymes and regulators, while metabolomics delivers quantitative measurements of pathway substrates, intermediates, and products. Their integration enables inference of active pathways and rate-limiting steps [83] [84].
The core premise is that coordinated changes in transcript levels for enzymes within a pathway often correlate with flux through that pathway, though post-translational regulation can decouple this relationship. Analytical validation therefore requires strategic experimental design and data integration approaches to accurately infer flux from multi-omics data.
Biosynthetic Building Blocks Context
In primary metabolism research, tracking flux from core building blocks such as acetyl-CoA, phosphoenolpyruvate, and amino acids into specialized metabolic pathways reveals how organisms prioritize resource allocation. Nematode-derived modular metabolites (NDMMs) exemplify this principle, where simple building blocks from primary metabolism—including dideoxysugars (ascarylose, paratose), lipid derivatives, amino acids, and neurotransmitters—are assembled into complex signaling architectures [81] [82]. Similar modular assembly principles operate in plant specialized metabolism, where primary metabolic precursors are diverted to secondary pathways under specific regulatory cues [85] [84].
Methodological Approaches
Experimental Design Considerations
Time-Series Sampling: Capturing metabolic dynamics requires strategic temporal design. Studies should include multiple time points spanning expected metabolic transitions, with sampling frequency determined by system kinetics. For example, Hâ‚‚Oâ‚‚ exposure experiments in fish muscle tissue employed 14-day exposure periods with sampling at multiple intermediates to track progressive metabolic changes [83].
Stimulus-Response Approaches: Perturbation experiments using substrates, inhibitors, or environmental changes reveal flux patterns by tracking system response. Nitrogen form experiments in Glycyrrhiza uralensis demonstrated how switching between ammonium and nitrate sources redirects flux between primary and secondary metabolism [84].
Multi-Tissue/Cellular Resolution: Spatial compartmentalization of metabolism necessitates tissue-specific or single-cell analyses. Advanced approaches now enable correlated single-cell RNA-seq and metabolomics from the same cells, providing unprecedented resolution for flux inference [85].
Metabolomics Methodologies
Liquid Chromatography-Mass Spectrometry (LC-MS):
- Extraction: Use pre-chilled ternary solvent (methanol/acetonitrile/water, 2:2:1) for comprehensive metabolite extraction [83].
- Chromatography: Employ reverse-phase and HILIC separations to cover diverse metabolite classes.
- Mass Analysis: High-resolution instruments (Orbitrap, Q-TOF) enable untargeted profiling and accurate mass measurement.
Data Processing:
- Peak detection and alignment using XCMS, MS-DIAL, or similar platforms
- Metabolite identification with spectral libraries (GNPS, HMDB)
- Relative quantification via peak area normalization
Transcriptomics Methodologies
RNA Sequencing:
- Library preparation: Strand-specific protocols preserve transcript orientation
- Sequencing depth: ≥30 million reads per sample for quantitative gene expression
- Platform options: Illumina dominates for cost-effectiveness and data quality
Differential Expression Analysis:
- Alignment: STAR, HISAT2 for mapping to reference genomes
- Quantification: FeatureCounts, HTSeq for read counting
- Statistical analysis: DESeq2, edgeR for identifying differentially expressed genes (DEGs)
Data Integration Strategies
Pathway-Centric Integration: Mapping both transcript and metabolite changes onto biochemical pathways (KEGG, MetaCyc) identifies coordinated changes. In carp muscle under Hâ‚‚Oâ‚‚ stress, integrated analysis revealed concordant changes in oxidative phosphorylation transcripts and TCA cycle metabolites [83].
Statistical Integration: Multivariate methods (canonical correlation analysis, O2PLS) identify latent variables explaining covariance between omics datasets.
Network-Based Approaches: Weighted gene co-expression network analysis (WGCNA) identifies gene modules whose expression profiles correlate with metabolite abundances, as demonstrated in aspen salicinoid biosynthesis research [85].
Experimental Protocols
Comprehensive Multi-Omics Workflow for Metabolic Flux Analysis
The following workflow outlines a standardized pipeline for generating and integrating transcriptomic and metabolomic data to infer metabolic flux.
Detailed Metabolomics Protocol
Sample Preparation:
- Homogenization: Tissue (100 mg) homogenized in 1 mL pre-chilled ternary solvent (methanol/acetonitrile/water, 2:2:1) using bead beater or mechanical homogenizer [83].
- Extraction: Vortex vigorously for 30 seconds, incubate at -20°C for 1 hour, centrifuge at 13,000× g for 15 minutes at 4°C.
- Storage: Transfer supernatant to MS vials, store at -80°C until analysis.
LC-MS Analysis:
- Chromatography: UHPLC system with HILIC and RP columns for comprehensive coverage.
- Mass Spectrometry: Data-dependent acquisition (DDA) and data-independent acquisition (DIA) modes on high-resolution mass spectrometer.
- Quality Control: Pooled quality control samples analyzed every 6-10 injections to monitor instrument performance.
Detailed Transcriptomics Protocol
RNA Extraction and Library Preparation:
- Extraction: Use TRIzol or column-based methods with DNase treatment.
- Quality Control: Assess RNA integrity (RIN > 8.0) using Bioanalyzer or TapeStation.
- Library Prep: Poly-A selection for mRNA, ribosomal RNA depletion for total RNA.
- Sequencing: Illumina platform, 150 bp paired-end reads, 30+ million reads per sample.
Bioinformatic Processing:
- Quality Control: FastQC for read quality assessment.
- Alignment: Map to reference genome using STAR or HISAT2.
- Quantification: Generate count matrices using featureCounts.
- Differential Expression: DESeq2 for statistical analysis of DEGs.
Data Analysis and Integration
Analytical Pathways for Multi-Omics Data Integration
The relationship between analytical approaches and their applications in metabolic flux research is summarized in the following diagram:
Key Analytical Outputs
Differential Expression and Abundance Analysis:
- Transcriptomics: Differentially expressed genes (DEGs) with statistical thresholds (FDR < 0.05, log2FC > 1)
- Metabolomics: Differentially abundant metabolites (FDR < 0.05, variable importance in projection > 1.5)
Pathway Enrichment Analysis:
- Gene set enrichment analysis (GSEA) for transcriptomic data
- Metabolite set enrichment analysis (MSEA) for metabolomic data
- Integrated pathway mapping using KEGG, Reactome, or PlantCyc databases
Correlation Networks:
- Pairwise correlation between metabolite abundances and transcript levels
- Identification of key regulator genes correlated with multiple pathway metabolites
Research Reagent Solutions
Table 1: Essential Research Reagents for Multi-Omics Metabolic Flux Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Extraction Solvents | Methanol/Acetonitrile/Water (2:2:1) | Comprehensive metabolite extraction for LC-MS analysis [83] |
| RNA Stabilization | TRIzol, RNAlater | Preserve RNA integrity during sample collection and storage |
| Chromatography Columns | HILIC, C18 reverse-phase | Separation of diverse metabolite classes prior to MS detection |
| Isotopic Tracers | ¹³C-glucose, ¹âµN-ammonium | Direct flux measurement through metabolic pathways |
| Library Prep Kits | Illumina TruSeq, NEBNext Ultra II | RNA library construction for sequencing |
| Reference Standards | Stable isotope-labeled internal standards | Metabolite quantification and instrument performance monitoring |
Case Studies and Applications
Nitrogen Metabolism and Specialized Metabolism in Plants
In Glycyrrhiza uralensis, integrated transcriptomics and metabolomics revealed how different nitrogen forms (ammonium vs. nitrate) redirect flux between primary and secondary metabolism. Ammonium nitrogen promoted growth and primary metabolism, while nitrate nitrogen enhanced flavonoid accumulation through coordinated upregulation of phenylpropanoid pathway genes and corresponding metabolite changes [84].
Table 2: Multi-Omics Analysis of Nitrogen Form Effects in G. uralensis
| Parameter | Ammonium Response | Nitrate Response |
|---|---|---|
| Growth Biomass | Significant increase | Moderate increase |
| Primary Metabolism | Enhanced amino acid biosynthesis, TCA cycle, glycolysis | Moderate enhancement of primary pathways |
| Secondary Metabolism | Moderate increase in flavonoids | Significant flavonoid accumulation |
| Key DEGs | Nitrogen assimilation genes (GS/GOGAT) | Phenylpropanoid pathway genes (PAL, CHS) |
| Regulatory Features | Coordinated upregulation of N uptake and assimilation | Redirected carbon flux to phenylpropanoids |
Oxidative Stress and Metabolic Dysregulation
In common carp muscle tissue, Hâ‚‚Oâ‚‚-induced oxidative stress caused significant metabolic dysregulation detected through integrated omics. Metabolomics identified 83 upregulated and 89 downregulated metabolites, predominantly lipids and organic acids, while transcriptomics revealed 470 upregulated and 451 downregulated genes enriched in muscle development and transcriptional regulation. Integrated analysis showed elevated oxidative phosphorylation and adipocytokine signaling pathways, demonstrating how environmental stress redirects metabolic flux [83].
Modular Metabolite Biosynthesis in Nematodes
Research on nematode-derived modular metabolites (NDMMs) exemplifies how tracking flux from primary metabolic building blocks reveals novel biochemical strategies. Ascaroside-based signaling molecules in C. elegans are assembled from dideoxysugar scaffolds (ascarylose) decorated with building blocks from lipid, amino acid, neurotransmitter, and nucleoside metabolism. This modular assembly strategy creates complex molecular architectures from simple primary metabolites, with multi-omics approaches essential for mapping the biosynthetic logic [81] [82].
Validation and Quality Control
Technical Validation
Metabolomics QC:
- Instrument stability: Pooled QC samples with coefficient of variation < 15%
- Retention time stability: < 0.1 min drift over sequence
- Mass accuracy: < 3 ppm error for known standards
Transcriptomics QC:
- RNA quality: RIN > 8.0
- Sequencing metrics: > 80% bases Q30, appropriate complexity
- Alignment rates: > 85% to reference genome
Biological Validation
Independent Validation:
- Enzyme activity assays for key pathway steps
- Isotopic tracer studies for direct flux confirmation
- Genetic manipulation (knockout/overexpression) to test predictions
Statistical Validation:
- Cross-validation of models for flux prediction
- Permutation testing to assess significance of correlations
- False discovery rate control for multiple testing
Integrated metabolomics and transcriptomics provides a powerful framework for analytical validation of metabolic flux in primary metabolism research. The methodologies outlined herein enable researchers to infer dynamic metabolic flows from static multi-omics measurements, revealing how organisms allocate primary metabolic building blocks to specialized metabolic pathways. As single-cell multi-omics and spatial metabolomics technologies advance, resolution of metabolic flux analysis will continue to improve, offering increasingly precise insights into metabolic regulation across biological systems.
Natural products (NPs) and specialized metabolites, derived from the building blocks of primary metabolism, are a vital source of bioactive compounds. A significant challenge in harnessing their potential lies in elucidating their biosynthetic pathways, which remain largely unknown for most compounds [60]. Computational methods, particularly those leveraging biosynthetic fingerprints, have emerged as powerful tools to address this gap. Unlike traditional molecular fingerprints designed for drug-like molecules, biosynthetic fingerprints explicitly encode structural features related to a compound's biosynthetic origin, providing a more interpretable and biologically relevant framework for pathway prediction and natural product classification [38] [86]. This technical guide details the core methodologies, experimental protocols, and key tools driving innovation in this field, framing the discussion within the broader context of biosynthetic building blocks from primary metabolism.
Core Concepts: From Building Blocks to Fingerprints
The biosynthesis of natural products is modular, originating from key primary metabolic pathways that supply universal building blocks [87]. These include:
- Acetate and Malonic Acid (AA/MA): For fatty acids, phenols, and polyketides.
- Mevalonic Acid or Methylerythritol Phosphate (MVA/MEP): For terpenoids and steroids, with the five-carbon isoprene units (IPP and DMAPP) as fundamental precursors.
- Shikimic Acid and Cinnamic Acid (CA/SA): For flavonoids, phenylpropanoids, and lignans.
- Amino Acids (AAs): For alkaloids and peptides [60].
Biosynthetic fingerprints capture the structural manifestations of these building blocks within the final natural product. Their design moves beyond purely structural characteristics to incorporate biosynthetic logic, thereby enhancing performance in tasks such as estimating biosynthetic similarity and predicting pathway origins [38].
Key Fingerprint Methodologies
Table 1: Comparison of Biosynthetic Fingerprint Approaches
| Fingerprint Name | Type | Key Features | Reported Performance Advantages |
|---|---|---|---|
| Biosynfoni [38] | Substructure Key (39 keys) | Based on biosynthetic building blocks from Dewick's biosynthetic logic; counted fingerprint; easily visualizable. | Outperforms MACCS, Morgan in biosynthetic distance estimation; comparable classification performance with higher interpretability. |
| Neural Fingerprints (GNNs) [86] | Learned Representation | Graph Neural Networks (GCN, GAT, GIN) learn features directly from molecular graph structures. | Outperform traditional, hand-crafted fingerprints in fine-grained NP classification tasks. |
| SubGrapher (SVMF) [88] | Visual Fingerprinting | Extracts functional groups and carbon backbones directly from molecular images, bypassing SMILES. | Superior retrieval performance and robustness for molecules and Markush structures in images. |
| MinHashed (MHFP) [87] | String-Based | Uses SMILES substrings as fragment identifiers, stored via the MinHash algorithm. | Effective for representing NPs in supervised bioactivity prediction tasks. |
Diagram 1: From primary metabolism to biosynthetic fingerprints.
Experimental Protocols and Methodologies
Protocol 1: Implementing and Validating a Substructure Key Fingerprint (Biosynfoni)
Objective: To create a biosynthesis-informed molecular fingerprint using a predefined set of biosynthetically relevant substructure keys and validate its performance in biosynthetic distance estimation and classification [38].
Materials & Reagents:
- Molecular Dataset: A collection of natural product structures in SMILES format (e.g., from COCONUT or LOTUS-DB).
- Cheminformatics Library: RDKit (or similar) for molecular handling and standard fingerprint generation.
- Computational Environment: Python programming environment with scientific computing stacks (NumPy, Scikit-learn).
Methodology:
- Substructure Key Definition: Define the set of 39 biosynthetic substructure keys based on established biosynthetic logic (e.g., from Dewick's book). Keys should represent building blocks from major pathways (acetate, shikimate, mevalonate, amino acids).
- Fingerprint Generation:
- For each molecule in the dataset, iterate through each substructure key.
- Check for the presence of the substructure within the molecule.
- Encode the result as a counted value (number of matches) for each key, generating a fixed-length integer vector.
- Biosynthetic Distance Validation:
- Identify a set of known biosynthetic reaction chains from databases like KEGG or MetaCyc.
- For compound pairs within a chain, calculate the Tanimoto similarity using the Biosynfoni fingerprint.
- Correlate the similarity scores with the number of enzymatic steps separating the pairs. A valid biosynthetic fingerprint should show a continuous decrease in similarity as the number of intervening steps increases.
- Classification Performance Benchmarking:
- Train a multi-label classifier (e.g., Random Forest) using the Biosynfoni fingerprint on a labeled natural product dataset (e.g., ChEBI classes).
- Benchmark performance against standard fingerprints (MACCS, Morgan) using metrics like F1-score and accuracy via cross-validation.
- Compare model training time and memory usage to evaluate computational efficiency.
Protocol 2: Predicting Biosynthetic Precursors using Machine Learning
Objective: To train a machine learning model to predict the primary metabolic precursors of plant-specialized metabolites [87].
Materials & Reagents:
- Curated Dataset: A dataset of specialized metabolites with annotated biosynthetic precursors (e.g., manually curated from KEGG, LOTUS-DB). Precursors include amino acids, GPP, FPP, GGPP, L-phenylalanine, etc.
- Fingerprint & Descriptor Set: A suite of molecular representations, including ECFP, MHFP, and biosynthetic fingerprints like Biosynfoni.
- Automated Machine Learning Engine: DeepMol AutoML or a similar framework to automate model selection and hyperparameter tuning.
Methodology:
- Data Preparation and Splitting:
- Compile and standardize molecular structures.
- Assign multi-labels based on known precursor relationships.
- Split the dataset into training (70%), validation (20%), and test (10%) sets. Consider alternative splits to test model robustness, such as excluding highly similar compounds from training.
- Model Training and Selection:
- Use an AutoML engine to train multiple multi-label classifiers (e.g., ridge classifiers, random forests, k-nearest neighbours) using different fingerprint inputs.
- Optimize hyperparameters over hundreds of trials using algorithms like the Tree-structured Parzen Estimator (TPE), with the goal of maximizing the macro F1-score on the validation set.
- Model Evaluation and Interpretation:
- Evaluate the best-performing pipeline on the held-out test set.
- Use metrics suitable for multi-label, unbalanced datasets, including macro F1 score (mF1), macro precision (mPrecision), and macro recall (mRecall).
- Analyze feature importance in linear models to identify which substructural elements are most predictive of specific precursors, enhancing interpretability.
Protocol 3: Multi-step Retrobiosynthetic Pathway Planning
Objective: To elucidate complete biosynthetic pathways for a target natural product from simple building blocks using deep learning and search algorithms [60].
Materials & Reagents:
- Reaction Database: A curated dataset of biochemical reactions (e.g., sourced from BioChem, MetaCyc, KEGG).
- Organic Reaction Data: A large dataset of organic reactions (e.g., USPTO) for data augmentation.
- Pathway Planning Tool: A tool like BioNavi-NP, which implements a retrobiosynthesis planning algorithm.
Methodology:
- Single-step Retrobiosynthesis Model Training:
- Train a transformer neural network model on a combined dataset of biochemical and NP-like organic reactions. This end-to-end model takes a product's SMILES as input and predicts potential precursor sets.
- Employ ensemble learning by combining multiple models to improve top-N prediction accuracy.
- Multi-step Pathway Planning:
- For a target NP, use the trained single-step model in an iterative, multi-step planning process.
- Implement a deep learning-guided AND-OR tree-based search algorithm (e.g., as in BioNavi-NP) to efficiently explore the combinatorial space of possible pathways from the target back to available building blocks.
- The algorithm evaluates pathways based on a cost function, prioritizing routes with higher likelihood.
- Pathway Validation and Enzyme Proposal:
- Assess the proposed pathways for their ability to recover known building blocks and reported pathways for test compounds.
- For each predicted biosynthetic step, use enzyme prediction tools (e.g., Selenzyme, E-zyme 2) to suggest plausible enzymes, facilitating experimental reconstruction.
Diagram 2: Retrobiosynthesis workflow.
Table 2: Key Computational Tools and Datasets for Biosynthetic Fingerprinting and Pathway Prediction
| Category | Item/Resource | Function/Description |
|---|---|---|
| Software & Libraries | RDKit | Open-source cheminformatics toolkit for fingerprint generation, substructure searching, and molecular manipulation. |
| DeepMol AutoML [87] | Automated machine learning engine for streamlining model selection and hyperparameter optimization for precursor prediction. | |
| PyTorch / TensorFlow | Deep learning frameworks for building and training custom neural network models, including GNNs. | |
| Databases | COCONUT [38] | A comprehensive collection of natural product structures for model training and validation. |
| LOTUS-DB [87] | A curated resource of natural products, useful for expanding precursor prediction studies. | |
| KEGG, MetaCyc [60] | Databases of biological pathways and enzymes, essential for curating training data and validating predicted pathways. | |
| Reactome [89] | Manually curated database of human biological pathways, useful for evaluating pathway prediction logic. | |
| Computational Tools | BioNavi-NP [60] | A navigable toolkit for predicting multi-step biosynthetic pathways using deep learning and tree-based search. |
| PathSingle [90] | A Python-based pathway analysis tool for single-cell data, demonstrating graph-based analysis of biological networks. | |
| Selenzyme / E-zyme 2 [60] | Tools for proposing plausible enzymes for a given biochemical reaction, complementing retrobiosynthesis predictions. |
Biosynthetic fingerprints represent a significant advancement over traditional molecular descriptors by embedding the logic of primary metabolism into the featurization of natural products. As detailed in this guide, methods ranging from interpretable substructure keys like Biosynfoni to powerful deep learning models like GNNs and BioNavi-NP are significantly improving our ability to classify natural products and predict their biosynthetic pathways. The integration of these computational aids, supported by robust experimental protocols and a growing toolkit of resources, is poised to accelerate the discovery and engineering of natural products for application in drug development and beyond.
Within the framework of biosynthetic building blocks derived from primary metabolism, the strategic selection and engineering of heterologous hosts have become a cornerstone of modern natural product research and development. Heterologous biosynthesis involves the transfer of biosynthetic gene clusters (BGCs) from their native producer into a surrogate host organism, thereby providing a viable route to access the beneficial properties of complex natural products that are often difficult to obtain through traditional extraction or chemical synthesis [91] [92]. This approach is particularly vital for compounds from marine microorganisms, a majority of which are uncultivable under standard laboratory conditions, leaving their vast biosynthetic potential untapped [93]. The success of this strategy, however, hinges on a critical comparative analysis of the available heterologous hosts and the engineering methodologies employed to optimize them. This review provides an in-depth technical evaluation of these hosts and approaches, offering a guide for their application in discovering and producing new generation therapeutics and biochemicals.
Heterologous Host Organisms: A Comparative Evaluation
The choice of a heterologous host is a foundational decision, profoundly influencing the success of pathway reconstitution and the yield of the target metabolite. The ideal host should be genetically tractable, easy to culture, and compatible with the expression of foreign BGCs, including their requisite post-translational modifications and substrate pools [94] [92]. The following section details the most commonly employed hosts, categorized by their phylogenetic and functional characteristics.
Table 1: Comparative Analysis of Common Heterologous Hosts
| Host Organism | Phylogenetic Class | Key Advantages | Key Disadvantages | Ideal for Natural Product Classes | Notable Production Example |
|---|---|---|---|---|---|
| Escherichia coli | Gram-negative Bacterium | Rapid growth, extensive genetic tools, simple cultivation, high recombinant protein yield [94] [91] | Lack of native precursors for some pathways, inability to perform eukaryotic PTMs, potential for protein misfolding and inclusion body formation [94] [95] | Type I, II, & III PKS, NRPS, Isoprenoids [91] | Aryl polyenes, diverse polyketides [91] |
| Streptomyces spp. | Actinobacterium (Gram-positive) | Native producers of many NPs, possess abundant secondary metabolite precursors, support expression of actinomycete BGCs with high fidelity [91] [92] | Slower growth than E. coli, more complex metabolism, less genetic tools available | PKS, NRPS, Hybrid PK-NRP [91] | Fredericamycin, Aminoglycosides [91] |
| Saccharomyces cerevisiae | Ascomycete (Fungus) | GRAS status, eukaryotic PTMs, strong genetic tools, capable of secreting proteins, facile homologous recombination [94] [96] | Hyperglycosylation of proteins, tough cell wall, low diversity of native secondary metabolites [94] | Terpenoids, Fatty Acid Derivatives, Polyketides [91] | Sesquiterpenes, Rubrofusarin [91] |
| Komagataella phaffii (Pichia pastoris) | Ascomycete (Fungus) | High biomass, strong inducible promoters (e.g., AOX1), Crabtree-negative, high protein secretion, GRAS status [94] [96] | Methanol requirement for induction, less genetic tools than S. cerevisiae | Recombinant Proteins, Peptides [96] [95] | Non-specific lipid-transfer proteins (nsLTP) [95] |
| Aspergillus spp. (e.g., A. nidulans, A. oryzae) | Filamentous Fungus | High secondary metabolite flux, can express large fungal BGCs, native-like environment for fungal enzymes [94] [97] | Complex background metabolism, potential for hazardous spores [94] | Fungal PKS, NRPS, RiPPs, Meroterpenoids [97] | Tenellin, Ilicicolin H, Sambutoxin [97] |
| Yarrowia lipolytica | Ascomycete (Fungus) | High secretory capacity, can utilize hydrophobic substrates, oleaginous [94] [96] | Less established as a NP production host | Lipases, Proteases, Terpenoids [96] [91] | Alpha-santalene, Homoeriodictyol [91] |
Prokaryotic Host Systems
Escherichia coli remains one of the most prevalent hosts due to its well-annotated genome, rapid growth in inexpensive media, and the availability of a vast arsenal of genetic manipulation tools [94] [91]. Its simplicity makes it an excellent chassis for expressing bacterial BGCs, particularly from Gram-negative bacteria. However, its inability to perform essential eukaryotic post-translational modifications (e.g., certain glycosylations) and its limited native pool of complex secondary metabolite building blocks (e.g., complex acyl-CoAs for polyketide biosynthesis) can pose significant hurdles [92]. Streptomyces species, being native prolific producers of secondary metabolites like polyketides and non-ribosomal peptides, offer a more specialized chassis. Their metabolism is naturally primed with essential precursors and cofactors, making them particularly suited for expressing large BGCs from other actinobacteria [91]. The main trade-offs are their slower growth rates and more complex genetic manipulation compared to E. coli.
Eukaryotic Host Systems
Eukaryotic hosts are indispensable for expressing BGCs from fungal or plant origins. Saccharomyces cerevisiae is a versatile host with robust molecular tools and the capacity for protein secretion and complex PTMs. Its status as "Generally Recognized As Safe" (GRAS) makes it attractive for pharmaceutical production [94] [96]. Komagataella phaffii is another methylotrophic yeast renowned for achieving very high cell densities and high-level recombinant protein production under the control of strong, inducible promoters like PAOX1 [96]. A comparative study on producing the hazelnut allergen Cor a 8 found that K. phaffii yielded a correctly folded, biologically active protein, whereas the E. coli-produced equivalent was misfolded and formed oligomers, highlighting the superiority of yeast for producing complex eukaryotic proteins [95]. Aspergillus species are filamentous fungi that serve as powerful hosts for reconstituting fungal natural product pathways. They provide a metabolic background rich in polyketide and non-ribosomal peptide precursors, which often leads to higher titers of the target fungal metabolite compared to other heterologous systems [97]. Their ability to handle large genomic DNA constructs and complex BGC regulation makes them ideal for mining cryptic fungal metabolism.
Engineering and Optimization Approaches for Heterologous Biosynthesis
The simple introduction of a BGC into a heterologous host is frequently insufficient for efficient production. Extensive host and pathway engineering are often required to achieve high yields and correct biosynthesis.
Genetic Manipulation and Pathway Refactoring
A critical first step is the successful cloning and assembly of the often-large BGCs (>10 kb). Advances in DNA assembly techniques, such as Gibson Assembly, Golden Gate cloning, and in vivo homologous recombination in yeast, have been pivotal [93] [96]. Refactoring—the process of reconstructing a BGC with synthetic genetic elements like strong native promoters, ribosome binding sites, and terminators—is a key strategy to bypass native regulatory hurdles and ensure strong, constitutive expression in the new host [97]. This was crucial in the unambiguous assignment of the sambutoxin BGC by expressing a refactored cluster from Fusarium oxysporum in Aspergillus nidulans [97].
Metabolic Engineering and Modeling
To channel the host's primary metabolic building blocks (e.g., acetyl-CoA, malonyl-CoA, amino acids) toward the heterologous pathway, metabolic engineering is essential. This involves:
- Overexpressing bottleneck enzymes in precursor supply pathways.
- Deleting or downregulating competing pathways that divert key intermediates.
- Implementing dynamic regulatory circuits to balance metabolic flux [94]. Computational modeling of metabolic networks can predict the availability of metabolites and identify optimal genetic modifications to support the heterologous pathway without compromising host viability [94].
Directed Evolution for Enzyme and Pathway Optimization
When rational design is limited by a lack of structural knowledge, directed evolution serves as a powerful complementary strategy. This method mimics natural evolution by employing iterative rounds of mutagenesis, screening, and amplification to steer enzymes or entire pathways toward a desired function [98] [99].
Table 2: Key Techniques in Directed Evolution
| Technique | Purpose | Key Advantage | Key Disadvantage |
|---|---|---|---|
| Error-Prone PCR | Introduce random point mutations across a gene [98] | Easy to perform; no prior structural knowledge needed [98] | Biased mutation spectrum; limited sampling of sequence space [98] |
| DNA Shuffling | Recombine sequences from multiple parent genes [98] [99] | Recombines beneficial mutations from different variants [99] | Requires high sequence homology between parents [98] |
| Site-Saturation Mutagenesis | Systematically randomize specific codons [98] | In-depth exploration of key residues; creates "smart" libraries [98] | Requires prior knowledge to select sites; libraries can become very large [98] |
| FACS-based Screening | Isolate variants based on fluorescence [98] | Extremely high throughput (millions of variants) [98] | Evolved property must be linked to a fluorescence change [98] |
The success of directed evolution hinges on the availability of a high-throughput screening assay to identify improved variants from large libraries. For enzymes, this often involves colorimetric or fluorogenic assays, or more sophisticated methods like fluorescence-activated cell sorting (FACS) [98]. Directed evolution has been successfully applied to improve enzyme stability, alter substrate specificity, and enhance the catalytic activity of biosynthetic enzymes expressed in heterologous hosts [99].
Experimental Workflows and Methodologies
A standardized workflow is essential for successful heterologous expression of natural product BGCs. The process can be broken down into key stages, from host selection to final compound characterization, with specific protocols for critical steps.
General Workflow for Heterologous Expression
The following diagram outlines the core iterative process of establishing and optimizing heterologous biosynthesis.
Diagram Title: Heterologous Biosynthesis Workflow
Detailed Key Experimental Protocols
Protocol 1: Golden Gate Assembly for BGC Refactoring This modular cloning method is highly efficient for assembling multiple DNA fragments simultaneously [96].
- Design: Inserts and the recipient vector are designed with Type IIS restriction enzyme sites (e.g., BsaI) that leave non-palindromic overhangs upon digestion.
- Digestion-Ligation: In a one-pot reaction, BsaI and T4 DNA Ligase are combined with the DNA parts. The enzyme cuts the sites, and the ligase joins the compatible overhangs.
- Transformation: The assembled plasmid is transformed into E. coli for propagation.
- Verification: Colonies are screened by colony PCR and plasmid sequencing to confirm correct assembly. The GoldenPiCs system is a specific implementation for K. phaffii [96].
Protocol 2: Functional Screening in Yeast using FACS For high-throughput screening of enzyme variants or production strains [98].
- Library Creation: Generate a library of variants via error-prone PCR or other mutagenesis methods and clone into a yeast expression vector.
- Transformation: Transform the library into S. cerevisiae to achieve a high representation of variants.
- Cultivation: Grow transformed yeast in microtiter plates or liquid culture.
- Assay & Sort: Employ a fluorogenic substrate or a product-specific fluorescent probe. Cells expressing enzymes with desired activity will become fluorescent. Use FACS to isolate the most fluorescent cell population.
- Recovery & Validation: Plate sorted cells to obtain single colonies and re-test individual clones for activity in a secondary assay.
Protocol 3: Metabolite Extraction and Analysis from Fungal Cultures For detecting and characterizing natural products from fungal hosts like Aspergillus nidulans [97].
- Fermentation: Grow the recombinant A. nidulans strain in appropriate liquid medium for 3-7 days at suitable temperature with agitation.
- Extraction: Separate mycelia and culture broth by filtration. Extract the broth with an equal volume of ethyl acetate. Extract the mycelia with methanol or acetone, then concentrate the solvent extracts in vacuo.
- Analysis: Resuspend the crude extract in methanol for analysis.
- LC-MS/HRMS: Use Liquid Chromatography coupled with High-Resolution Mass Spectrometry to separate metabolites and determine their accurate mass. Compare the mass and UV spectrum to standards if available.
- NMR: For structural elucidation of novel compounds, purify the metabolite using preparatory HPLC or flash chromatography and analyze by 1D and 2D Nuclear Magnetic Resonance spectroscopy.
The Scientist's Toolkit: Essential Research Reagents and Solutions
The following table details key reagents, materials, and tools essential for conducting heterologous biosynthesis experiments.
Table 3: Essential Research Reagents and Solutions
| Tool/Reagent | Function/Description | Application Example |
|---|---|---|
| antiSMASH Software | A bioinformatics platform for the automated identification and analysis of biosynthetic gene clusters in genomic data [93]. | Primary analysis of sequenced microbial or metagenomic DNA to locate candidate BGCs for heterologous expression [93]. |
| Golden Gate MoClo Kit | A modular cloning system based on Type IIS restriction enzymes that allows for the assembly of multiple DNA parts in a single reaction [96]. | Refactoring and assembling large BGCs into expression vectors for hosts like E. coli, yeast, or Aspergillus [96]. |
| pPICZA Vector (for K. phaffii) | An expression vector containing the AOX1 promoter for strong, methanol-inducible expression and a Zeocin resistance marker for selection [96] [95]. | Secretory production of recombinant proteins and peptides in Komagataella phaffii [95]. |
| ChromAzurol S (CAS) Assay | A colorimetric assay used to detect siderophores (iron-chelating compounds) [93]. | Functional screening of metagenomic libraries constructed in E. coli for the heterologous production of novel siderophores [93]. |
| Fluorogenic Substrate Probes | Synthetic substrate molecules that release a fluorescent signal upon enzymatic cleavage or modification. | High-throughput screening of enzyme variant libraries generated by directed evolution using microplate readers or FACS [98]. |
The strategic selection and engineering of heterologous hosts provide an indispensable platform for accessing the chemical diversity of natural products, firmly built upon the foundation of primary metabolism. As this comparative analysis demonstrates, no single host is universally superior; the choice depends on a careful balance of the BGC's origin, complexity, and required post-translational modifications, against the host's genetic tractability, metabolic capacity, and scalability. The continued development of synthetic biology tools, CRISPR-based genome editing, and sophisticated metabolic models will further enhance our ability to engineer these biological factories. By leveraging the distinct advantages of each host system and applying a combination of refactoring, metabolic engineering, and directed evolution, researchers can systematically unlock the vast potential of cryptic biosynthetic pathways, accelerating the discovery and development of next-generation drugs and fine chemicals.
The translation of primary metabolism research into viable biosynthetic building blocks represents a cornerstone of modern industrial biotechnology. This process involves engineering microbial cell factories to function as chemical plants, harnessing endogenous metabolic pathways for the production of high-value compounds, from pharmaceuticals to specialty chemicals [100]. However, the journey from a laboratory-scale proof-of-concept to an economically viable industrial process requires meticulous benchmarking and optimization. The complexity of biological systems introduces unique challenges in scaling, including metabolic burden, precursor flux limitations, and host toxicity, which are not encountered in traditional chemical manufacturing [101] [100]. Consequently, the success of any biomanufacturing process is critically dependent on a framework of Key Performance Indicators (KPIs) that quantitatively bridge the gap between cellular physiology and industrial operational excellence. This guide provides a comprehensive overview of these essential metrics, detailing their calculation, application, and significance in de-risking the scale-up of biosynthetic processes.
Core Key Performance Indicators for Biomanufacturing
Effective benchmarking requires a multi-faceted approach to performance measurement. The following tables categorize and define the essential KPIs for industrial-scale biosynthetic production, integrating classic manufacturing metrics with biology-specific parameters.
Table 1: Core Production and Efficiency KPIs
| KPI | Formula | Application in Biosynthesis |
|---|---|---|
| Throughput | # of Units Produced / Time | Measures the production capability of a bioreactor or production line over a specified period (e.g., mg/L/hour) [102]. |
| Titer | Mass of Product / Volume of Broth (g/L) | The final concentration of the target compound in the fermentation broth; a primary indicator of pathway efficiency and host performance [103]. |
| Yield | Mass of Product / Mass of Substrate (g/g) | Indicates the carbon conversion efficiency from the raw material (e.g., glucose) to the desired product, critical for cost-effectiveness [103]. |
| Productivity (Rate) | Titer / Fermentation Time (g/L/h) | Reflects the speed of the production process, integrating both titer and time, which directly impacts facility throughput [103]. |
| Right First Time (RFT) | (Units Produced Correctly First Time / Total Units Produced) * 100 | Percentage of product batches meeting quality specifications without rework; indicates process robustness and control [102]. |
| Cycle Time | Process End Time – Process Start Time | The total time required to complete one production batch, from inoculation to harvest [102]. |
Table 2: Metabolic and Cellular Performance KPIs
| KPI | Formula / Description | Significance |
|---|---|---|
| Specific Productivity | (Titer) / (Cell Density * Time) (pg/cell/day) | Measures the production efficiency of each individual cell, distinguishing between high titer from high cell density versus superior pathway function [101]. |
| Precursor Carbon Conversion | (Moles of Carbon in Product / Moles of Carbon in Consumed Substrate) * 100 | Quantifies the metabolic flux diverted from central carbon metabolism (e.g., MVA or MEP pathways) into the target biosynthetic pathway [100]. |
| Metabolic Burden | Measured as reduction in host growth rate (μ) or biomass yield upon pathway induction. | Indicates the fitness cost imposed by the heterologous pathway; a lower burden is essential for stable industrial fermentations [101]. |
| ATP/Redox Co-factor Balance | Theoretical vs. Actual consumption of ATP, NADPH, etc. | Identifies potential co-factor limitations that can create bottlenecks in the engineered pathway [101]. |
Table 3: Operational and Economic KPIs
| KPI | Formula | Interpretation |
|---|---|---|
| Overall Equipment Effectiveness (OOE) | Availability * Performance * Quality | A holistic analysis of production efficiency, accounting for availability (downtime), performance (speed), and quality (yield of good product) [102]. |
| Inventory Turns | Cost of Goods Sold / Average Inventory | Measures supply chain efficiency. High turns indicate effective resource use and low risk of raw material degradation [102]. |
| Avoided Cost | (Assumed Repair Cost + Production Losses) – Preventive Maintenance Cost | Estimates savings from preventive actions (e.g., prophylactic equipment maintenance or genetic stabilization of the production host) [102]. |
| Return on Assets (ROA) | (Net Income / Total Assets) * 100 | Evaluates how efficiently the company is using its assets (including bioreactor capacity) to generate profit [102]. |
Experimental Protocols for KPI Determination
Accurate KPI determination relies on standardized experimental methodologies. The following protocols are essential for generating reliable and comparable data.
Protocol for Determining Production Titer, Yield, and Rate
This protocol outlines the procedure for quantifying the three most critical production metrics in a batch fermentation system.
- Inoculum Preparation: Inoculate a single colony of the engineered production strain into a seed culture medium. Grow overnight to reach mid- to late-exponential phase.
- Bioreactor Operation: Inoculate the main bioreactor containing the production medium to a specified initial optical density (OD600). Precisely record the initial volume and substrate concentration (e.g., glucose).
- Process Control: Maintain critical environmental parameters (temperature, pH, dissolved oxygen) at setpoints throughout the fermentation. For induced systems, add the inducer at the specified cell density.
- Sampling: Aseptically withdraw samples at defined intervals (e.g., every 2-4 hours) for analysis.
- Analytical Measurements:
- Cell Density: Measure OD600 or cell dry weight (CDW) for each sample.
- Substrate Concentration: Quantify the concentration of the primary carbon source (e.g., using HPLC or a bioanalyzer).
- Product Concentration: Quantify the target product concentration using validated analytical methods (e.g., LC-MS, GC-MS, HPLC).
- Data Calculation:
- Titer (g/L): The product concentration measured at the end of the fermentation.
- Yield (Yp/s, g/g): (Final Titer - Initial Titer) / (Initial Substrate Concentration - Final Substrate Concentration).
- Productivity (g/L/h): Maximum Titer / Total Fermentation Time.
Protocol for Assessing Metabolic Burden and Specific Productivity
This protocol quantifies the physiological impact of the heterologous pathway on the host organism.
- Strain Cultivation: Cultivate the production strain and an isogenic control strain (lacking the production pathway) in parallel under identical conditions, both with and without pathway induction.
- Growth Kinetics: Monitor OD600 frequently to generate high-resolution growth curves. Calculate the maximum specific growth rate (μmax) for each condition by fitting the exponential phase data to the equation: ln(OD) = ln(OD0) + μmax * t.
- Cell-Specific Analysis: At key timepoints, measure both the product titer and the cell density (as CDW).
- Data Calculation:
- Metabolic Burden: Calculate the percentage reduction in μmax: [(μmaxcontrol - μmaxproduction) / μmax_control] * 100.
- Specific Productivity: Calculate at the time of peak production: (Product Titer) / (CDW * Fermentation Duration).
Visualizing the Biosynthetic Workflow: From Design to KPIs
The path to successful industrial production is an iterative cycle of design, construction, testing, and learning. The following diagram visualizes this workflow and the points at which different KPIs are applied.
The Scientist's Toolkit: Essential Research Reagent Solutions
The experimental workflow relies on a suite of critical reagents and tools to design, construct, and analyze production strains.
Table 4: Key Research Reagents and Tools for Biosynthetic Engineering
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| Retrobiosynthesis Software | Predicts novel enzymatic pathways to a target molecule from available precursors [104] [105]. | Tools like RetroPath2.0 are used in the Design Phase to explore potential routes to a plant natural product [103] [105]. |
| Metabolic Databases | Provide curated information on compounds, reactions, enzymes, and pathways across species [104]. | KEGG and MetaCyc are used for Pathway Design and Host Selection by identifying enzyme sequences and verifying pathway feasibility [104] [100]. |
| Cell-Free Expression Systems | A lysate-based platform for rapid protein synthesis and pathway prototyping without living cells [106]. | Used in the Test Phase to express and assay enzyme variants quickly, bypassing the need for lengthy in vivo transformations [107] [106]. |
| Analytical Standards | Highly purified reference compounds for instrument calibration and quantification. | Essential for KPI Calculation (Titer, Yield) via HPLC or LC-MS to ensure accurate measurement of the target product and metabolic intermediates [100]. |
| Strain Engineering Kits | Modular cloning toolkits (e.g., BioBricks, Golden Gate) for standardized DNA assembly [103]. | Utilized in the Build Phase to rapidly assemble multiple gene expression cassettes for the heterologous pathway into the production host [103]. |
Conclusion
The strategic exploitation of biosynthetic building blocks from primary metabolism represents a cornerstone of modern drug discovery and sustainable pharmaceutical production. By integrating foundational knowledge of metabolic pathways with advanced methodological approaches in synthetic biology, researchers can overcome historical limitations of yield and complexity. The continued development of sophisticated troubleshooting and validation tools, including machine learning-powered biosynthetic fingerprints and precise genome editing, is rapidly accelerating our capacity to engineer nature's chemical logic. Future directions will likely focus on creating increasingly intelligent and automated platforms for pathway design, further bridging the gap between laboratory discovery and clinical application to address pressing global health challenges with novel, naturally-inspired therapeutics.
References
- 1. Secondary metabolism is not secondary—why we need more ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12394244/]
- 2. Secondary Metabolites in Plants [https://www.mdpi.com/2223-7747/14/14/2146]
- 3. Microbial secondary metabolites: advancements to ... [https://pubmed.ncbi.nlm.nih.gov/39824928/]
- 4. Systems and synthetic biology for plant natural product ... [https://www.sciencedirect.com/science/article/pii/S2211124725004863]
- 5. Plant Metabolites: primary vs. secondary | Research Starters [https://www.ebsco.com/research-starters/anatomy-and-physiology/plant-metabolites-primary-vs-secondary]
- 6. Primary vs Secondary Metabolites- Definition, 12 ... [https://microbenotes.com/primary-vs-secondary-metabolites/]
- 7. Production of secondary metabolites under challenging ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1569014/full]
- 8. ß-NAD serving as a building for natural product block ... biosynthesis [https://communities.springernature.com/posts/ss-nad-serving-as-a-building-block-for-natural-product-biosynthesis-a-novel-link-between-primary-and-secondary-metabolism]
- 9. Frontiers | Editorial: Essence of survival: impact of primary and... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1651327/full]
- 10. The shikimate pathway: gateway to metabolic diversity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11043010/]
- 11. Synthesis, Building Blocks, Energy - Metabolism [https://www.britannica.com/science/metabolism/The-synthesis-of-building-blocks]
- 12. Identification of the key metabolites and related genes ... [https://www.sciencedirect.com/science/article/pii/S096399692201345X]
- 13. Deep learning driven biosynthetic pathways navigation for ... [https://www.nature.com/articles/s41467-022-30970-9]
- 14. A computational framework to explore large-scale biosynthetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6917865/]
- 15. A community resource for paired genomic and ... [https://www.nature.com/articles/s41589-020-00724-z]
- 16. A Streamlined Metabolic Pathway For the Biosynthesis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1936435/]
- 17. Research Briefing in 2025 | Nature Metabolism [https://www.nature.com/natmetab/articles?type=research-briefing&year=2025]
- 18. phenolics, terpenes, and alkaloids [https://www.sciencedirect.com/science/article/abs/pii/S0981942824003425]
- 19. Origin and Function of Structural Diversity in the Plant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8621143/]
- 20. Biotechnological production of terpenoids using cell factories [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1621406/full]
- 21. Advances in the Biosynthesis of Plant Terpenoids [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114759/]
- 22. Discovery and characterisation of terpenoid biosynthesis ... [https://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-025-06421-0]
- 23. Alkaloid biosynthesis in medicinal crop kratom (Mitragyna ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1653916/full]
- 24. Biotechnological production of terpenoids using cell factories [https://pmc.ncbi.nlm.nih.gov/articles/PMC12568664/]
- 25. The Biosynthesis of Phenolic Compounds Is an Integrated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7765139/]
- 26. sciencedirect.com/topics/engineering/ primary -metabolite [https://www.sciencedirect.com/topics/engineering/primary-metabolite]
- 27. and secondary metabolites and primary in natural... building blocks [https://studyx.ai/questions/4lgcokr/primary-and-secondary-metabolites-and-building-blocks-in-natural-product]
- 28. From data to discovery: leveraging big data in plant natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12183336/]
- 29. Omics strategies for plant natural product biosynthesis [https://www.maxapress.com/article/id/68477d20fa6c5854b3e1f387]
- 30. Hierarchical metabolic engineering for rewiring cellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12514951/]
- 31. Toward Engineering Synthetic Microbial Metabolism - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2796345/]
- 32. Advanced applications of synthetic biology technology in ... [https://www.sciencedirect.com/science/article/pii/S167463842500125X]
- 33. Pathway enrichment analysis and visualization of omics data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6607905/]
- 34. Analysis Tools [https://reactome.org/userguide/analysis]
- 35. Combinatorial biosynthesis for the engineering of novel ... [https://www.nature.com/articles/s42004-024-01172-9]
- 36. Reinvigorating natural product combinatorial biosynthesis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4757526/]
- 37. Comparing total chemical synthesis and total biosynthesis ... [https://pubs.rsc.org/en/content/articlehtml/2025/np/d4np00015c]
- 38. Biosynfoni: a biosynthesis-informed and interpretable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12395878/]
- 39. Shining a light on enzyme promiscuity [https://www.sciencedirect.com/science/article/abs/pii/S0959440X17300234]
- 40. Enzyme Promiscuity: Engine of Evolutionary Innovation [https://pmc.ncbi.nlm.nih.gov/articles/PMC4215207/]
- 41. A recursive pathway for isoleucine biosynthesis arises from ... [https://elifesciences.org/reviewed-preprints/108864]
- 42. An Evolutionary Biochemist's Perspective on Promiscuity [https://pmc.ncbi.nlm.nih.gov/articles/PMC4836852/]
- 43. Enzyme promiscuity [https://en.wikipedia.org/wiki/Enzyme_promiscuity]
- 44. Enzyme promiscuity prediction using hierarchy-informed ... [https://arxiv.org/abs/2002.07327]
- 45. Enzyme promiscuity in the field of synthetic biology applied ... [https://www.sciencedirect.com/science/article/pii/S2693125725000275]
- 46. Frustration can Limit the Adaptation of Promiscuous ... [https://link.springer.com/article/10.1007/s00239-024-10161-4]
- 47. Advanced metabolic engineering strategies for increasing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10898619/]
- 48. Single-nucleus transcriptomics reveal the morphogenesis ... [https://www.nature.com/articles/s41467-025-63770-y]
- 49. Artemisinin: a game-changer in malaria treatment [https://www.mmv.org/malaria/symptoms-and-treatments/treatments-about-ACTs]
- 50. Malaria: Artemisinin partial resistance [https://www.who.int/news-room/questions-and-answers/item/artemisinin-resistance]
- 51. Artemisinin production strategies for industrial scale [https://www.sciencedirect.com/science/article/abs/pii/S0926669024009142]
- 52. Approaches and Recent Developments for the Commercial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5797932/]
- 53. Safeguarding artemisinin: integrating bioproduction ... [https://malariajournal.biomedcentral.com/articles/10.1186/s12936-025-05648-4]
- 54. Method and device for the synthesis of artemisinin [https://patents.google.com/patent/EP2565197A1/en]
- 55. Validation of ELISA for Quantitation of Artemisinin-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3854889/]
- 56. Integrated metabolomic and transcriptomic analysis ... [https://www.sciencedirect.com/science/article/abs/pii/S0141813025085551]
- 57. A novel fed-batch cultivation strategy based on tyrosine ... [https://www.sciencedirect.com/science/article/abs/pii/S0168165625002494]
- 58. Combinatorial engineering pinpoints shikimate pathway ... [https://jbioleng.biomedcentral.com/articles/10.1186/s13036-025-00553-5]
- 59. Advancing lignocellulosic conversion though biosensor ... [https://pubs.rsc.org/en/content/articlehtml/2025/gc/d5gc03618f]
- 60. Deep learning driven biosynthetic pathways navigation for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9187661/]
- 61. Uncoupling protein production from growth [https://pmc.ncbi.nlm.nih.gov/articles/PMC12519851/]
- 62. Unveiling Metabolic Engineering Strategies by Quantitative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11615770/]
- 63. Resource-allocation constraint governs structure and ... [https://www.sciencedirect.com/science/article/pii/S1096717621001968]
- 64. Questions, data and models underpinning metabolic ... [https://www.frontiersin.org/journals/systems-biology/articles/10.3389/fsysb.2022.998048/full]
- 65. Synthetic biology strategies for microbial biosynthesis of ... [https://www.nature.com/articles/s41467-019-09848-w]
- 66. Precision Metabolic Engineering: the Design ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4770000/]
- 67. Crosstalk between primary and secondary metabolism [https://pmc.ncbi.nlm.nih.gov/articles/PMC11239236/]
- 68. Explain enzyme specificity (lock and key model) [https://www.sparkl.me/learn/cambridge-igcse/biology-0610-core/explain-enzyme-specificity-lock-and-key-model/revision-notes/3739]
- 69. An enzyme activation network reveals extensive regulatory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12222706/]
- 70. Integrative genomic mining for enzyme ... | Nature Communications [https://www.nature.com/articles/ncomms10005?error=cookies_not_supported&code=b6551f50-e376-42e4-ab05-a8141f1c8ab9]
- 71. Gene networks that compensate for crosstalk with crosstalk [https://www.nature.com/articles/s41467-019-12021-y]
- 72. Advances in Protein Engineering for Enzyme in... Optimization [https://www.linkedin.com/pulse/advances-protein-engineering-enzyme-optimization-biopharma-mcfbf]
- 73. Towards a Synthetic Biology Toolset for Metallocluster Enzymes in... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8617995/]
- 74. Frontiers | CRISPR and RNA : revolutionary tools for... interference [https://www.frontiersin.org/journals/food-science-and-technology/articles/10.3389/frfst.2025.1609948/full]
- 75. RNAi vs. CRISPR: Guide to Selecting the Best Gene ... [https://www.synthego.com/blog/rnai-vs-crispr-guide/]
- 76. Gene Silencing – RNAi or CRISPR? [https://nordicbiosite.com/blog/gene-silencing-rnai-or-crispr]
- 77. Novel CRISPR -Cas Systems: An Updated Review of the Current ... [https://pubmed.ncbi.nlm.nih.gov/33805113/]
- 78. Evaluation of RNAi and CRISPR technologies by large ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5726721/]
- 79. Evolution-guided optimization of biosynthetic pathways - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4273373/]
- 80. RNAi-mediated control of CRISPR functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7295050/]
- 81. Modular assembly of primary metabolic building blocks [https://pmc.ncbi.nlm.nih.gov/articles/PMC4304883/]
- 82. Modular Assembly of Primary Metabolic Building Blocks [https://www.sciencedirect.com/science/article/pii/S1074552114003743]
- 83. Integrated Metabolomics and Transcriptomics Reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12466421/]
- 84. Transcriptomics and metabolomics reveal the primary and ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2023.1229253/full]
- 85. A Metabolomics and Transcriptomics Resource for Identifying ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12508522/]
- 86. Leveraging Molecular Graphs for Natural Product ... [https://www.sciencedirect.com/science/article/pii/S2001037025003538]
- 87. Predicting precursors of plant specialized metabolites using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12569576/]
- 88. Subgrapher: visual fingerprinting of chemical structures [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01091-4]
- 89. Evaluating the predictive accuracy of curated biological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9216552/]
- 90. Pathway metrics accurately stratify T cells to their cells states [https://biodatamining.biomedcentral.com/articles/10.1186/s13040-024-00416-7]
- 91. Heterologous biosynthesis as a platform for producing new ... [https://www.sciencedirect.com/science/article/abs/pii/S0958166920300872]
- 92. Methods and options for the heterologous production of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9896020/]
- 93. Recent Advances in the Heterologous Expression of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9225448/]
- 94. Heterologous Metabolic Pathways: Strategies for Optimal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7385092/]
- 95. Pichia pastoris is superior to E. coli for the production of ... [https://www.sciencedirect.com/science/article/abs/pii/S1046592809002149]
- 96. Comparison of Yeasts as Hosts for Recombinant Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6027275/]
- 97. Deciphering Chemical Logic of Fungal Natural Product ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9906657/]
- 98. A primer to directed evolution: current methodologies and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10074555/]
- 99. Directed evolution [https://en.wikipedia.org/wiki/Directed_evolution]
- 100. Engineering microbial cell factories for the production of plant natural... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-017-0732-7]
- 101. Constructing de novo biosynthetic pathways for chemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3768262/]
- 102. 40+ Manufacturing KPIs & Metrics for 2025 | insightsoftware [https://insightsoftware.com/blog/30-manufacturing-kpis-and-metric-examples/]
- 103. Automated engineering of synthetic metabolic pathways for ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717620301828]
- 104. computational methods for biosynthetic pathway design [https://pmc.ncbi.nlm.nih.gov/articles/PMC12159830/]
- 105. Planning biosynthetic pathways of target molecules based ... [https://www.sciencedirect.com/science/article/abs/pii/S147692712400094X]
- 106. Cell-free synthetic biology for natural product biosynthesis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11920963/]
- 107. Biosynthetic Methods in Pharmaceutical Manufacturing [https://www.biopharminternational.com/view/biosynthetic-methods-in-pharmaceutical-manufacturing]