CRISPR-Guided Evolution: Accelerating Protein Engineering and Drug Discovery
This article provides a comprehensive guide for researchers on CRISPR-Cas mediated directed evolution, a transformative methodology for accelerating protein engineering.
CRISPR-Guided Evolution: Accelerating Protein Engineering and Drug Discovery
Abstract
This article provides a comprehensive guide for researchers on CRISPR-Cas mediated directed evolution, a transformative methodology for accelerating protein engineering. We explore the foundational principles of coupling CRISPR-Cas systems with directed evolution workflows, detailing key methodological protocols for gene diversification, screening, and selection. The guide addresses common troubleshooting and optimization challenges, compares CRISPR-based approaches to traditional evolution methods, and validates success through case studies in enzyme engineering, antibody development, and therapeutic protein optimization. Finally, we discuss future implications for streamlining drug discovery pipelines.
From Natural Evolution to Lab Acceleration: Core Principles of CRISPR-Directed Evolution
Application Notes
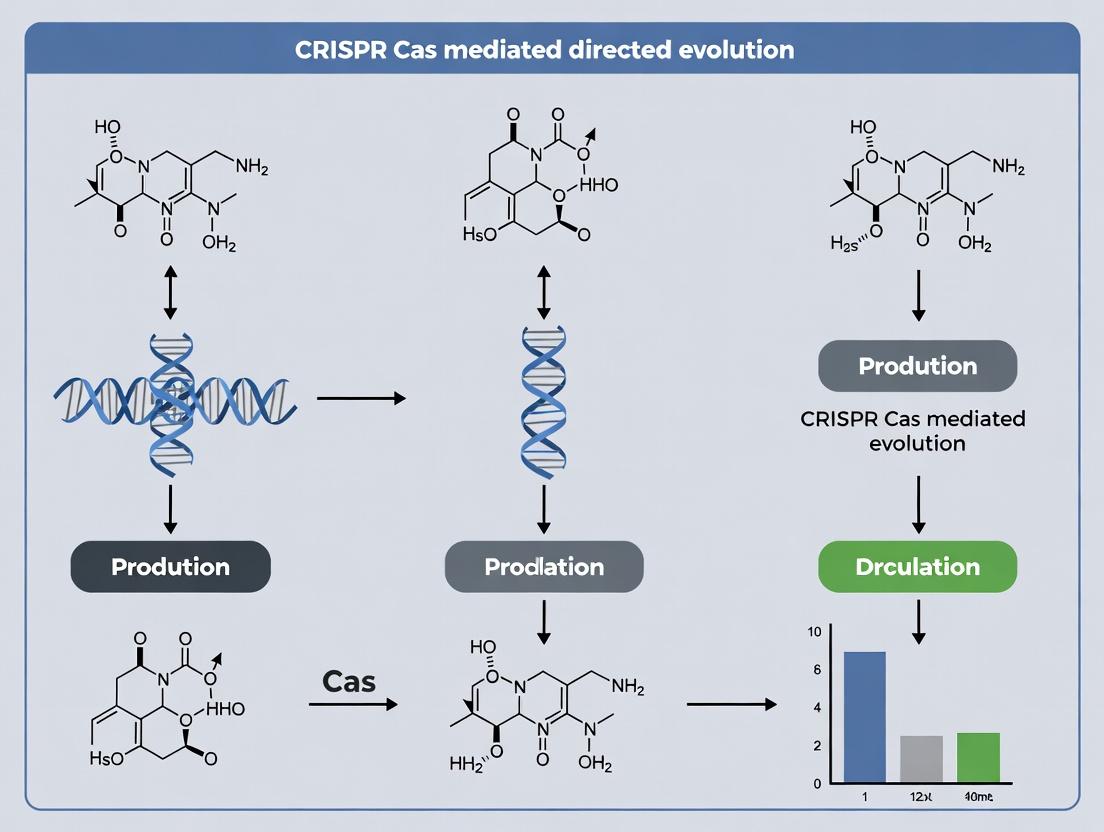
CRISPR-Cas mediated directed evolution (CMDE) represents a transformative integration of adaptive cellular machinery with iterative phenotypic selection. This approach leverages the precision of CRISPR systems to generate and link genetic diversity to selectable cellular outcomes, dramatically accelerating the evolution of proteins with enhanced or novel functions. Within the broader thesis of CRISPR-Cas directed evolution research, this methodology is posited as a unifying framework that moves beyond random mutagenesis and low-throughput screening.
The core principle involves using a CRISPR-Cas system, typically Cas9 or Cas12a, to introduce targeted double-strand breaks (DSBs) in a gene of interest (GOI) within a living cell. The cell's subsequent repair, primarily via error-prone non-homologous end joining (NOMEJ) or homology-directed repair (HDR) with mutagenic donor libraries, creates a diverse mutant pool in situ. Crucially, the genotype (variant DNA) remains physically linked to its phenotype (encoded protein function) within the cell, enabling direct selection or screening (e.g., for antibiotic resistance, fluorescence, binding affinity, or enzymatic activity under pressure). Selected cells are then harvested, and the enriched mutant sequences can be identified via next-generation sequencing (NGS).
Key Advantages and Quantitative Benchmarks
The quantitative superiority of CMDE over traditional methods is evident in several metrics:
Table 1: Performance Comparison of Directed Evolution Platforms
| Metric | Traditional Methods (e.g., Error-Prone PCR) | CRISPR-Cas Mediated Directed Evolution |
|---|---|---|
| Library Size (Variants) | 10^6 - 10^8 (in vitro) | 10^7 - 10^10 (in vivo) |
| Mutation Rate (per kb) | 1-20 (random, global) | Tunable, 1-100+ (targeted, local) |
| Selection Throughput | Low to medium (often requires separate screening) | Very high (direct phenotypic coupling) |
| Cycle Time (Days) | 7-14 | 3-5 |
| Genotype-Phenotype Linkage | Artificial (e.g., phage/yeast display) | Natural (within the host cell) |
Table 2: Representative CMDE Achievements in Protein Engineering
| Protein Target | Evolved Trait | Fold Improvement/Result | CRISPR System Used |
|---|---|---|---|
| TEM-1 β-lactamase | Antibiotic Resistance (Ceftazidime) | >100-fold increase in MIC | Cas9-NOMEJ |
| GFP | Fluorescence Intensity | 20-fold enhancement | Cas12a-HDR |
| Anti-PD1 scFv | Binding Affinity (KD) | 5 nM to 50 pM (100x) | Cas9 with ssDNA donor library |
| Cytosine Deaminase | Targeting Specificity | 10x reduced off-target editing | Base Editor directed evolution |
Protocols
Protocol 1: CMDE via Cas9-Mediated NOMEJ for Antibiotic Resistance Evolution
Objective: To evolve enhanced antibiotic resistance in a bacterial β-lactamase gene.
Workflow Diagram:
Title: CMDE via Cas9 and NHEJ Workflow
Detailed Methodology:
- Construct Design: Clone the GOI (e.g., blaTEM-1) into a plasmid co-expressing Cas9 and a specific sgRNA targeting within the GOI.
- Library Preparation: Transform the construct into E. coli (end-joining proficient strains like MG1655 ΔrecA may be used).
- Diversity Generation: Induce Cas9/sgRNA expression with anhydrotetracycline (aTc, 100 ng/mL) for 2-4 hours to generate DSBs. Allow repair via native error-prone NOMEJ for 16-24 hours.
- Selection: Plate cells on LB agar containing a gradient (or fixed high concentration) of the target antibiotic (e.g., ceftazidime from 2 µg/mL to 64 µg/mL). Incubate at 37°C for 24-48 hours.
- Enrichment & Sequencing: Pick surviving colonies from the highest antibiotic concentration. Pool, isolate plasmid DNA, and prepare amplicons of the GOI for NGS (Illumina MiSeq, 2x300 bp).
- Analysis: Align sequences to the wild-type GOI, identify mutation spectra and enriched variants. Reclone top hits for validation.
Protocol 2: CMDE via Cas9/dCas9-Mediated Targeted Mutagenesis with HDR
Objective: To evolve a mammalian cell surface receptor for improved ligand binding using a dCas9-cytidine deaminase fusion and a donor oligonucleotide library.
Pathway/System Diagram:
Title: Targeted Mutagenesis with Base Editor & HDR
Detailed Methodology:
- Cell Line Engineering: Stably express dCas9-APOBEC1 (a cytidine deaminase) and the target receptor in a mammalian cell line (e.g., HEK293T).
- Library Delivery: Transfect cells with a pool of sgRNAs targeting the receptor gene's extracellular domain and a complex library of single-stranded donor oligonucleotides (ssODNs, 100-200 nt) containing degenerate codons at specified positions.
- Mutation Generation: The dCas9-deaminase complex localizes and creates C-to-T (or G-to-A) transitions. The ssODN library serves as a template for HDR, introducing further diversity.
- Selection: After 72-96 hours, label cells with a fluorescently-tagged ligand. Use fluorescence-activated cell sorting (FACS) to isolate the top 1-5% of cells with the highest fluorescence (indicating highest binding).
- Recovery & Iteration: Expand sorted cells. Genomic DNA is extracted, the target region is amplified by PCR, and the process (steps 2-4) is repeated for 3-5 rounds.
- Analysis: Clone final PCR products and sequence individual clones, or perform bulk NGS to identify consensus mutations.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for CMDE
| Reagent/Material | Function in CMDE | Example/Notes |
|---|---|---|
| Cas9/dCas9 Expression Vector | Provides the DNA-cleaving or DNA-binding scaffold. | pCas9 (Addgene #42876), pX458 (Addgene #48138). |
| sgRNA Library Cloning System | Enables multiplexed targeting of the GOI. | Lentiguide-Puro (Addgene #52963) or custom array synthesis. |
| Mutagenic Repair Template Library | Introduces targeted diversity via HDR. | Ultramer DNA Oligos (IDT) with NNK/C degenerate codons. |
| Error-Prone Repair Proficient Host | Facilitates NOMEJ-mediated mutagenesis. | E. coli MG1655 ΔrecA ΔendA strains. |
| Selection Agent | Applies phenotypic pressure to enrich functional variants. | Antibiotics, fluorescent ligands, FACS antibodies, toxin metabolites. |
| NGS Library Prep Kit | Enables high-throughput analysis of variant libraries. | Illumina Nextera XT, Swift Accel-NGS 2S Plus. |
| Base/Double Base Editor Plasmid | Enables precise, single-nucleotide diversification without DSBs. | pCMV_BE3 (Addgene #73021) for C-to-T. |
| HDR Enhancer Chemical | Increases HDR efficiency for donor template incorporation. | RS-1 (Rad51 stimulator), Scr7 (Ligase IV inhibitor). |
Classical directed evolution mimics natural selection by introducing genetic diversity (typically via random mutagenesis or gene recombination) followed by screening or selection for desired traits. Modern genome editing, particularly CRISPR-Cas systems, provides precise, targeted genetic modifications. This synergy creates a powerful paradigm for accelerated protein and cellular engineering. Within CRISPR-Cas mediated directed evolution research, the core thesis is that CRISPR systems can be engineered to not just edit, but to continuously and diversely evolve genomic loci in a targeted, continuous, and high-throughput manner, thereby bridging the scale of classical methods with the precision of modern editing.
Application Note 1: CRISPR-Cas Mediated Continuous Evolution (MAGE-CRISPR) This approach combines multiplex automated genome engineering (MAGE) with CRISPR-Cas targeting to enable rapid, iterative cycles of diversification and selection in living cells, such as E. coli or yeast. It is ideal for evolving metabolic pathways or protein complexes.
Application Note 2: Targeted Diversity Generation with Base Editors & Prime Editors CRISPR base editors (BEs) and prime editors (PEs) enable precise, single-nucleotide diversification at defined genomic loci without requiring double-stranded breaks or donor templates. This is applied for probing protein function via saturated mutagenesis or evolving gain-of-function alleles.
Application Note 3: In Vivo Mutagenesis with Error-Prone CRISPR-Cas Fusion of a error-prone DNA polymerase or deaminase domain to a nicking Cas9 variant (e.g., nCas9) creates a localized hypermutator. This continuously introduces mutations within a window around the target site, simulating classical random mutagenesis but with locus-specific control.
Protocols
Protocol 1: CRISPR-Cas Mediated Phage-Assisted Continuous Evolution (PACE) for Protein Engineering Objective: Evolve a protein-of-interest (POI) through continuous selection in bacterial host cells using a CRISPR-modified phage propagation system. Workflow:
- Construct Host Cell Strain: Engineer an E. coli host cell to express: a) a mutagenesis plasmid (e.g., expressing a nCas9-APOBEC1 deaminase fusion for targeted C-to-T diversity), b) a selection plasmid where phage propagation is dependent on the desired activity of the evolving POI.
- Construct Phage Vector: Clone the gene for the POI into an accessory plasmid (AP) packaged into M13 phage particles. The AP must lack the gene for the essential phage protein pIII, whose expression is made dependent on POI function in the host.
- Initiate Evolution: Infect the host cell culture with the engineered phage in a continuous flow chemostat (lagoon). Fresh host cells flow in, and phage outflow is monitored.
- Selection Pressure: Only phage that have acquired beneficial mutations in the POI gene that enhance the activity triggering pIII expression will propagate and be carried out in the effluent.
- Harvest & Analysis: Sample effluent phage daily. Sequence the POI gene from phage DNA to track evolution. Typical PACE runs last 7-10 days, achieving 10s-100s of generations. Key Parameters: Flow rate (1-2 host cell doublings per hour), mutagenesis rate (tuned by promoter strength of mutagenesis plasmid), and selection stringency.
Protocol 2: Saturation Mutagenesis of a Protein Domain Using CRISPR-Base Editor Libraries Objective: Create and screen all possible single amino acid substitutions within a specific protein domain. Workflow:
- Design sgRNA Library: Design a tiled library of sgRNAs targeting every codon within the genomic region encoding the protein domain. For a 100-amino acid domain, design ~100 sgRNAs.
- Clone sgRNA Library: Clone the pooled sgRNA library into a lentiviral vector backbone.
- Generate BE-expressing Cell Line: Stably express a base editor (e.g., BE4max) in your mammalian cell line of interest.
- Transduce & Edit: Transduce the BE-expressing cells with the sgRNA lentiviral library at a low MOI to ensure single integrations. Culture cells for 7-10 days to allow editing.
- Apply Selection/FACS: Apply relevant drug or functional selection, or sort cell populations based on a desired phenotype using FACS.
- Deep Sequencing & Analysis: Extract genomic DNA from pre-selection and post-selection populations. Amplify the target region and sequence via NGS. Enrichment scores for each sgRNA/variant are calculated.
Table 1: Quantitative Comparison of Directed Evolution Platforms
| Platform | Typical Mutation Rate | Diversity Type | Throughput (Library Size) | Key Application | Cycle Time |
|---|---|---|---|---|---|
| Error-Prone PCR (Classical) | 1-20 mutations/kb | Random, global | 10⁶ - 10¹¹ | Enzyme activity improvement | Weeks |
| CRISPR-Cas PACE | 10⁻⁵ - 10⁻³ mutations/bp/gen | Targeted, continuous | Continuous (>10¹² over run) | Protein-protein interactions, catalysis | Days (continuous) |
| CRISPR-BE Saturation | >90% editing efficiency per target base | Targeted, single nucleotide | 10² - 10⁵ sgRNAs per gene | Functional mapping, drug resistance studies | 2-3 weeks |
| Prime Editing Saturation | Variable (10-50% efficiency) | Targeted, small insertions/deletions | 10³ - 10⁵ pegRNAs | All possible substitutions & indels | 3-4 weeks |
Visualizations
Title: Evolution of Directed Evolution Techniques
Title: CRISPR-Cas PACE System Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Function & Explanation |
|---|---|
| nCas9 (D10A) - APOBEC1 Fusion Plasmid | Expresses a nickase Cas9 fused to a cytidine deaminase. Creates targeted C-to-T (or G-to-A) mutations without double-strand breaks, essential for in vivo hypermutation. |
| Lentiviral Base Editor (BE4max) System | High-efficiency base editor for mammalian cells. Enables stable integration and expression, allowing for large-scale, pooled sgRNA library screens with consistent editing. |
| Pooled sgRNA or pegRNA Library | A synthesized DNA library containing thousands of unique guide RNAs targeting a gene or region. The diversity driver for saturation mutagenesis screens. |
| M13 Phage Accessory Plasmid (AP) | Engineered phage plasmid lacking essential genes (e.g., pIII). Serves as the vector for the evolving gene of interest during PACE experiments. |
| Chemostat/Lagoon Apparatus | A continuous-flow bioreactor that maintains constant cell growth conditions. Critical for PACE, allowing for the continuous influx of fresh hosts and outflow of evolved phage. |
| FACS Aria or Equivalent Cell Sorter | Fluorescence-activated cell sorter. Enables high-throughput isolation of mammalian cells based on phenotypic changes (e.g., fluorescence, surface markers) resulting from editing. |
| Next-Generation Sequencing (NGS) Kit | For deep sequencing of target genomic loci pre- and post-selection. Essential for quantifying variant enrichment and identifying beneficial mutations. |
| Selection Circuit Plasmid (for PACE) | Plasmid encoding the genetic logic that links the desired activity of the protein-of-interest to the expression of an essential gene for phage propagation (e.g., pIII). The engine of selection pressure. |
Within the broader thesis of CRISPR-Cas mediated directed evolution research, this Application Note delineates the core mechanistic principles that enable these systems to generate targeted genetic diversity and directly couple it to selectable phenotypes. This foundational capability allows researchers to accelerate evolutionary trajectories for protein engineering, metabolic pathway optimization, and therapeutic discovery.
Core Mechanisms and Application Notes
Mechanism of Targeted Diversity Generation
CRISPR-Cas systems, particularly nuclease-deactivated variants (dCas), are engineered to recruit mutagenic agents to specific genomic loci. This targeted approach contrasts with random mutational methods, concentrating diversity in user-defined regions of interest (e.g., a specific gene promoter or protein-coding sequence).
Key Application Note: The fusion of dCas9 to activation-induced cytidine deaminase (AID) or error-prone DNA polymerases creates a targeted diversity generator. For example, the fusion protein dCas9-PMCD1 (a plant-derived cytidine deaminase) enables C•G to T•A transitions at a high frequency within a narrow window (~35-65 bp) from the protospacer adjacent motif (PAM).
Mechanism of Phenotype Coupling
The generated genetic diversity remains physically linked to the encoding DNA within the cell. This intrinsic link ensures that a genotype conferring a beneficial phenotype (e.g., antibiotic resistance, fluorescence, growth advantage) can be selectively enriched and its sequence identified through next-generation sequencing.
Key Application Note: Continuous evolution systems like EvolvR and VEGAS integrate the diversity generation module directly into the host genome. Cells that undergo beneficial mutations are immediately selected for, and their mutated plasmids or genomic loci are harvested for analysis, creating a seamless genotype-to-phenotype link.
Table 1: Performance Metrics of Key CRISPR-Cas Diversity Generation Systems
| System Name | Core Fusion/Component | Mutation Type Generated | Typical Mutation Rate (vs. background) | Targeting Window | Primary Application |
|---|---|---|---|---|---|
| Target-AID | dCas9 + pmCDA1 (AID) | C→T (G→A) | 10⁻³ to 10⁻⁵ (≥100x) | ~35-65 bp from PAM | Bacterial & yeast protein engineering |
| EvolvR | nCas9 (D10A) + error-prone Pol I | All base substitutions | 10⁻⁵ to 10⁻⁷ (≥1,000x) | Tunable, ~70 bp | Continuous evolution in E. coli |
| CRISPR-X | dCas9 + MS2-AID | C→T, G→A | ~0.1% per base (≥100x) | ~100 bp window | Mammalian cell protein evolution |
| VEGAS | dCas9 + Activation-induced AID (AID) | C→T, G→A | Not quantified (High) | Transcriptional start site | Signaling pathway engineering in mammalian cells |
Table 2: Phenotype Coupling Efficiency in Recent Studies (2023-2024)
| Study Focus | CRISPR-DE System Used | Selection Pressure | Enrichment Factor (Mutant/WT) | Key Identified Mutant | Ref. |
|---|---|---|---|---|---|
| Antibody Affinity Maturation | dCas9-AID variant | Flow cytometry (antigen binding) | ~500x | Fab variant with 40x improved KD | Lee et al., 2023 |
| TEM-1 β-lactamase Evolution | EvolvR | Ceftazidime (antibiotic) | >10,000x | TEM-1 with 4 new mutations conferring resistance | Shivram et al., 2024 |
| GFP Fluorescence Enhancement | Targeted CRISPR-X | FACS (fluorescence) | ~200x | GFP with 2.5x increased brightness | Zhao et al., 2023 |
Detailed Experimental Protocols
Protocol 4.1: Targeted Diversity Generation Using a dCas9-AID System inE. coli
Objective: Introduce targeted C-to-T mutations within a specific gene of interest.
Materials:
- E. coli strain expressing dCas9-AID fusion protein from a plasmid.
- Second plasmid expressing guide RNA (gRNA) targeting the gene of interest.
- Target plasmid containing the gene to be evolved.
- LB media with appropriate antibiotics (e.g., carbenicillin, chloramphenicol).
- SOC outgrowth media.
Procedure:
- Transformation: Co-transform the dCas9-AID plasmid and the gRNA plasmid into competent E. coli cells already harboring the target plasmid. Plate on LB agar with all three required antibiotics. Incubate at 37°C overnight.
- Diversity Generation Culture: Pick 5-10 colonies and inoculate a 5 mL starter culture. Dilute 1:100 into 5 mL of fresh LB with antibiotics and 1 mM IPTG (to induce dCas9-AID expression). Grow for 16-24 hours at 30°C (slower growth allows more mutation cycles).
- Harvest and Pool: Pellet the cells. Extract the pool of target plasmids using a miniprep kit. This plasmid pool now contains a library of targeted mutations.
- Analysis: Transform the plasmid pool into a fresh, reporter E. coli strain for phenotypic selection or subject to next-generation sequencing to assess mutation spectrum and frequency.
Protocol 4.2: Phenotype-Coupled Continuous Evolution Using EvolvR
Objective: Evolve a gene for a new function under continuous selection without iterative cloning.
Materials:
- E. coli strain with genomic integration of EvolvR (nCas9-errPol I).
- Guide RNA plasmid targeting the genomic locus of the gene to be evolved.
- Selection plates or media (e.g., containing increasing antibiotic concentration).
- PCR primers flanking the target locus.
Procedure:
- Setup: Transform the gRNA plasmid into the EvolvR E. coli strain. Plate on selective media.
- Continuous Evolution Passage: Inoculate a single colony into liquid media with antibiotic and 0.2% arabinose (to induce EvolvR expression). Grow to saturation (~24 hrs).
- Apply Selection: Plate a portion of the saturated culture onto solid media containing the desired selection pressure (e.g., a high concentration of antibiotic). Also, perform a serial dilution and plate on non-selective media to determine total viable cell count.
- Iterate and Isolate: Pick colonies from the selection plate. Use these to inoculate fresh liquid media for the next round of growth and selection. Repeat for 3-10 passages.
- Genotype Analysis: After significant enrichment is observed (e.g., growth under selective conditions), perform colony PCR on selected clones using the flanking primers. Sequence the amplicons to identify accumulated mutations.
Visualization Diagrams
Title: CRISPR-Cas Targeted Diversity Generation Workflow
Title: Phenotype Coupling Logic in Cellular Selections
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for CRISPR-Cas Directed Evolution Experiments
| Item | Function in Experiment | Example Product/Catalog Number (Representative) |
|---|---|---|
| dCas9-AID Fusion Plasmid | Expresses the core targeting and mutagenesis machinery. | Addgene #113864 (pEvolvR-dCas9-AID) |
| Guide RNA (gRNA) Expression Plasmid | Directs the Cas fusion to the specific DNA target locus. | Custom designed, cloned into backbone like pTargetF. |
| Error-Prone Polymerase Fusion Plasmid | For systems like EvolvR; provides broad mutational spectrum. | Addgene #124369 (pEvolvR-NG) |
| Chemically Competent E. coli Cells | Essential for library transformation and propagation. | NEB 5-alpha or similar; also specialized strains like MG1655 mutS-. |
| Next-Generation Sequencing Kit | For deep sequencing of mutant libraries to assess diversity. | Illumina DNA Prep Kit. |
| Fluorescence-Activated Cell Sorter (FACS) | For high-throughput phenotypic selection based on fluorescence. | Instrument: BD FACSAria. |
| Selection Antibiotics | To maintain plasmid pressure and apply phenotypic selection. | Carbenicillin, Chloramphenicol, Kanamycin. |
| Inducers (IPTG, Arabinose) | To precisely control the expression timing/level of CRISPR-DE components. | Isopropyl β-d-1-thiogalactopyranoside (IPTG), L-Arabinose. |
1. Application Notes
Within directed evolution research, CRISPR tools enable the rapid generation of diverse, targeted genotype-phenotype linkages in living cells, accelerating the exploration of fitness landscapes. The evolution of these tools from simple cutters to precise editors and modulators underpins modern in vivo directed evolution platforms.
- Cas9 Nuclease (First-Generation): Serves as the foundational tool for creating double-strand breaks (DSBs), which are repaired via error-prone non-homologous end joining (NHEJ) to generate random indel libraries at target genomic loci. It is ideal for functional knockout screens and creating pools of diverse, disruptive variants for evolutionary selection pressures.
- Base Editors (BEs; Second-Generation): Catalyze direct, irreversible chemical conversion of one DNA base into another without inducing DSBs. Cytosine Base Editors (CBEs) enable C•G to T•A transitions. Adenine Base Editors (ABEs) enable A•T to G•C transitions. Their high efficiency and low indel rates are optimal for introducing all possible transition mutations across a gene of interest to model evolutionary trajectories and refine protein function.
- Prime Editors (PEs; Third-Generation): Function as "search-and-replace" tools, capable of installing all 12 possible base-to-base conversions, as well as small insertions and deletions, with minimal byproducts. Their precision and versatility allow for the systematic introduction of specific, pre-determined allelic series found in natural populations or hypothesized beneficial combinations, testing evolutionary hypotheses.
- CRISPR Activation/Interference (CRISPRa/i): Utilize a catalytically dead Cas9 (dCas9) fused to transcriptional effectors (e.g., dCas9-VPR for activation; dCas9-KRAB for interference). These tools modulate gene expression levels without altering the underlying DNA sequence, enabling artificial selection on transcriptional programs and mimicking regulatory evolution in high-throughput screens.
2. Quantitative Data Summary
Table 1: Comparison of Key CRISPR Tools for Directed Evolution
| Tool | Core Component | Primary Editing Outcome | Typical Efficiency Range* | Indel Byproduct Rate* | Key Advantage for Evolution Studies |
|---|---|---|---|---|---|
| Cas9 Nuclease | Cas9 + gRNA | Random indels (NHEJ) | 20-80% (indels) | N/A | Simplicity; generates diverse, disruptive mutation spectrum. |
| Cytosine Base Editor | dCas9/nCas9 + cytidine deaminase + gRNA | C•G to T•A transitions | 10-50% (avg. product) | 0.1-10% | High efficiency, low indels; models transition mutations. |
| Adenine Base Editor | dCas9/nCas9 + adenosine deaminase + gRNA | A•T to G•C transitions | 10-40% (avg. product) | 0.1-5% | High efficiency, low indels; models complementary transitions. |
| Prime Editor | nCas9-H reverse transcriptase + PE gRNA | All point mutations, small insertions/deletions | 5-30% (avg. product) | <1-5% | Versatility & precision; installs specific haplotypes. |
| CRISPRa | dCas9 + transcriptional activator + gRNA | Gene expression upregulation | Varies (2-100x induction) | N/A | Selects on phenotype from tunable expression levels. |
| CRISPRi | dCas9 + transcriptional repressor + gRNA | Gene expression knockdown | Varies (50-90% knockdown) | N/A | Selects on phenotype from tunable expression knockdown. |
*Efficiencies are highly dependent on cell type, delivery, and target locus.
3. Experimental Protocols
Protocol 1: Multiplexed Cas9 Nuclease Screening for Drug Resistance Variants Objective: To generate and select for genetic variants conferring resistance to a targeted therapeutic agent.
- Library Design: Design a pool of sgRNAs tiling the exons of the target gene (e.g., BTK for BTK inhibitors). Clone into a lentiviral sgRNA expression vector.
- Virus Production: Produce high-titer lentivirus in HEK293T cells using standard packaging plasmids.
- Cell Infection & Selection: Infect target cells (e.g., leukemic cell lines) at a low MOI (<0.3) to ensure single integrations. Select with puromycin for 72 hours.
- Variant Enrichment: Apply the selective pressure (e.g., Ibrutinib) to the library population. Maintain for 2-4 weeks, passaging as needed.
- Genomic DNA Extraction & Sequencing: Harvest genomic DNA from pre-selection and post-selection populations. Amplify the sgRNA cassette by PCR and subject to next-generation sequencing (NGS).
- Analysis: Compare sgRNA abundance pre- and post-selection using MAGeCK or similar algorithms to identify enriched guides and inferred resistant variants.
Protocol 2: Saturation Base Editing for Functional Mapping Objective: To assess the fitness consequence of all possible transition mutations within a protein domain.
- BE Selection: Choose an appropriate CBE (e.g., BE4max) or ABE (e.g., ABEmax) based on the desired base conversion.
- gRNA Library Design: Design a library of gRNAs spaced to place every cytidine or adenosine within the target window (positions 4-10, R-loop) within the protospacer of at least one guide.
- Delivery: Co-deliver the BE plasmid and the gRNA library plasmid pool via nucleofection into the target cell line.
- Harvest & Sorting: After 72-96 hours, harvest cells. For surface proteins, stain with a fluorescently labeled antibody and sort populations with high, medium, and low expression via FACS.
- Targeted Amplicon Sequencing: Isolate genomic DNA from sorted populations. Perform PCR amplification of the target region and sequence via NGS.
- Analysis: Use software (e.g, BE-Analyzer) to calculate the frequency of each base conversion in each population. Fitness scores are derived from the enrichment/depletion of specific edits across sorted bins.
4. Visualizations
Diagram Title: Cas9 Screening for Evolved Drug Resistance
Diagram Title: CRISPR Tool Functions in Directed Evolution
5. The Scientist's Toolkit
Table 2: Essential Research Reagents for CRISPR-directed Evolution
| Reagent / Material | Function in Evolution Context |
|---|---|
| Lentiviral sgRNA/Editor Constructs | Stable delivery and integration of CRISPR machinery for long-term selection experiments. |
| Chemically Defined sgRNA Library | Defines the targeted mutational space (e.g., gene-wide, domain-specific). Critical for pool screening. |
| High-Efficiency Transfection Reagent (e.g., Nucleofector) | Enables delivery of editor RNP or plasmid to hard-to-transfect primary or stem cells. |
| Puromycin/Blasticidin/Other Selection Agents | Selects for cells successfully transduced with the CRISPR vector during library establishment. |
| Phenotypic Selection Agent (e.g., Drug, Cytokine) | Applies the evolutionary pressure to enrich for desired genetic variants. |
| FACS Aria or Similar Cell Sorter | Isolates cell populations based on complex phenotypic readouts (e.g., surface marker, reporter fluorescence). |
| NGS Library Prep Kit (for Amplicon Seq) | Prepares the amplified target genomic regions from pooled populations for deep sequencing. |
| Analysis Software (MAGeCK, BE-Analyzer, CRISPResso2) | Computationally identifies enriched guides or quantifies editing outcomes from NGS data. |
Application Notes
Within a thesis on CRISPR-Cas mediated directed evolution, the Central Dogma provides the conceptual framework linking designed genetic perturbations (genotype) to measurable cellular outcomes (phenotype). High-throughput CRISPR screening operationalizes this link for functional genomics and therapeutic target discovery. The integration of next-generation sequencing (NGS) quantifies genotype abundance, creating a powerful, quantitative readout for evolutionary selection or phenotypic fitness.
Key Quantitative Metrics in CRISPR Screening: The success and quality of a screen are evaluated using standardized metrics. The following table summarizes critical quantitative benchmarks.
Table 1: Key Quantitative Data and Benchmarks for Pooled CRISPR Screens
| Metric | Typical Target Value | Description & Importance |
|---|---|---|
| Library Coverage | > 200x per sgRNA | Read depth ensuring each sgRNA is adequately sampled in the plasmid library. |
| Cell Coverage | > 500x per sgRNA | Number of transduced cells per sgRNA to minimize stochastic dropout effects. |
| Transduction Efficiency | 30-60% | Percentage of cells expressing the Cas9/sgRNA; ensures population-level representation. |
| Screen Performance (Pearson R²) | > 0.8 (for replicates) | Correlation between biological replicates indicates high reproducibility. |
| Hit Identification (FDR / p-value) | FDR < 0.05, p < 0.01 | Statistical thresholds for identifying significantly enriched/depleted sgRNAs/genes. |
| Gene Effect Score (e.g., CERES, MAGeCK) | Variable (e.g., < -0.5 for essential) | Normalized score quantifying gene knockout effect on fitness. Negative = depletion. |
Experimental Protocols
Protocol 1: Pooled CRISPR-knockout Screening for Essential Genes Objective: To identify genes essential for cell proliferation/survival under standard culture conditions.
Library Design & Preparation:
- Utilize a genome-scale lentiviral sgRNA library (e.g., Brunello, 4 sgRNAs/gene).
- Amplify the plasmid library via ultra-deep sequencing (min. 200x coverage) to verify representation.
Cell Line Preparation:
- Maintain Cas9-expressing cells (or stably transduce) in appropriate medium. Confirm >90% Cas9 activity via flow cytometry or surrogate reporter assay.
Lentiviral Transduction & Selection:
- Transduce cells at a low MOI (~0.3) to ensure most cells receive only one sgRNA. Include a non-targeting control sgRNA pool.
- At 24-48 hours post-transduction, add selection antibiotic (e.g., Puromycin, 1-5 µg/mL) for 3-7 days to eliminate untransduced cells.
Harvesting Timepoints for Genomic DNA (gDNA):
- T0 Harvest: Collect a minimum of 5e6 cells (maintaining >500x coverage) immediately after selection. Pellet, wash with PBS, and store at -80°C.
- Tfinal Harvest: Culture the remaining population for ~14 population doublings. Harvest a minimum of 5e6 cells as above.
gDNA Extraction & sgRNA Amplification:
- Extract gDNA using a mass-scale kit (e.g., Qiagen Blood & Cell Culture Maxi Kit). Ensure yield >50 µg.
- Perform a two-step PCR to amplify integrated sgRNA cassettes from gDNA and attach Illumina sequencing adapters/indexes. Use a minimum of 100 µg gDNA per sample to maintain library complexity.
Sequencing & Analysis:
- Pool PCR products and sequence on an Illumina platform (MiSeq/HiSeq). Aim for >200x coverage of the original library.
- Align reads to the sgRNA library reference. Use analysis pipelines (MAGeCK, CRISPResso2) to calculate sgRNA depletion/enrichment and gene-level significance scores (e.g., MAGeCK RRA).
Protocol 2: CRISPRa/i Screening for Drug Resistance Phenotypes Objective: To identify gene activations (CRISPRa) or repressions (CRISPRi) that confer resistance to a chemotherapeutic agent.
Library & Cell Line:
- Use a targeted lentiviral sgRNA library designed for dCas9-VPR (activation) or dCas9-KRAB (interference).
- Use a cell line stably expressing the appropriate dCas9-effector protein.
Transduction & Selection:
- Follow Protocol 1, Steps 2-3, to generate a pooled, selected cell population.
Perturbation & Selection:
- Split cells into two arms: DMSO Control and Drug-Treated.
- Treat cells with the drug at a pre-determined IC70-IC80 concentration. Maintain cultures, passaging as needed, for 14-21 days.
- Harvest cell pellets (5e6-10e6 cells) from both arms at the endpoint.
Downstream Processing & Hit Calling:
- Extract gDNA and prepare sequencing libraries as in Protocol 1, Step 5-6.
- Analyze sequencing data to identify sgRNAs significantly enriched in the drug-treated arm compared to the control, indicating a resistance-conferring perturbation.
Visualizations
Title: Workflow for Pooled CRISPR-Cas9 Screening
Title: Central Dogma in CRISPR Screening
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Pooled CRISPR Screening
| Reagent / Material | Function & Brief Explanation |
|---|---|
| Genome-scale sgRNA Library (e.g., Brunello, GeCKO) | Pre-designed, pooled collection of sgRNA plasmids targeting all known genes. Provides the genetic perturbation source. |
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Second-generation system for producing recombinant lentivirus to deliver the sgRNA and selection marker. |
| Stable Cas9/dCas9-Effector Cell Line | Engineered cells constitutively expressing the nuclease (Cas9) or programmable activator/repressor (dCas9-VPR/KRAB). Essential for consistent editing. |
| Polybrene (Hexadimethrine Bromide) | A cationic polymer that enhances viral transduction efficiency by neutralizing charge repulsion between virus and cell membrane. |
| Puromycin (or other antibiotics) | Selective agent for cells successfully transduced with the lentiviral vector, which contains a resistance gene. |
| Mass gDNA Extraction Kit | Scalable kit for isolating high-quality, high-quantity genomic DNA from millions of pooled screening cells. |
| High-Fidelity PCR Master Mix | For accurate, minimal-bias amplification of the integrated sgRNA cassettes from gDNA prior to sequencing. |
| Illumina Sequencing Platform & Reagents | Provides the high-throughput, quantitative readout of sgRNA abundance in the population before and after selection. |
| Bioinformatics Pipeline (MAGeCK, CRISPResso2) | Software for aligning sequencing reads, counting sgRNAs, and performing statistical analysis to identify significant hits. |
Building Your Pipeline: Step-by-Step Protocols and Real-World Applications
Application Notes: A CRISPR-Cas Mediated Directed Evolution Framework
Within a thesis on CRISPR-Cas mediated directed evolution, this protocol provides a systematic pipeline for accelerating protein or functional nucleic acid evolution. This approach integrates targeted mutagenesis with phenotypic selection, bypassing the need for extensive library construction and screening. The core innovation lies in using CRISPR-Cas systems to introduce diversity in situ and link genotype to phenotype within living cells, enabling continuous evolution cycles.
Key Workflow Modules
1. Target Gene Selection & gRNA Design Selection criteria are paramount. Ideal candidates possess quantifiable phenotypes (e.g., fluorescence, survival, binding affinity) and are amenable to mutational drift without lethal effects.
- Metrics for Selection: Demonstrated functional plasticity in nature, availability of a high-throughput screen or selection, and defined functional domains.
- gRNA Design: Design 2-3 gRNAs targeting regions proximal to functional domains but avoiding essential catalytic residues to maintain baseline function. Tools like CHOPCHOP or Benchling are used with specificity checks against the host genome.
Table 1: Quantitative Parameters for Target Gene Selection
| Parameter | Optimal Range | Measurement Method |
|---|---|---|
| Gene Length | 0.5 - 3 kb | Sequencing |
| Baseline Activity | >10% of wild-type | Functional assay (e.g., enzymatic rate) |
| Number of gRNAs | 2-3 per gene | In silico design tools |
| gRNA On-target Efficiency | >70% relative activity | T7E1 or NGS assay |
| gRNA Off-target Score | <60 (CCTop) | In silico prediction |
2. CRISPR-Cas Mutagenesis System Integration The chosen system dictates the mutation profile.
- Base Editors (BE): For precise point mutation libraries (C•G to T•A or A•T to G•C).
- Prime Editors (PE): For targeted insertions, deletions, and all 12 possible point mutations.
- Cas9 with Error-Prone Repair: Co-delivery of Cas9, gRNA, and an error-prone DNA repair template (e.g., using low-fidelity polymerases) for localized diversity.
- Orthogonal Systems: Using Cas12a for multiplexing or nickase versions to reduce indels.
Protocol 1: Lentiviral Delivery of Base Editor & Selection Cassette Objective: Stably integrate the mutagenesis machinery and a survival gene (e.g., antibiotic resistance) linked to the target gene's function.
- Clone: Insert the target gene, a programmable promoter (e.g., tetracycline-inducible), and a downstream antibiotic resistance gene (e.g., puromycin) into a lentiviral transfer plasmid.
- Package: Co-transfect HEK293T cells with the transfer plasmid, psPAX2 (packaging), and pMD2.G (envelope) plasmids using PEI transfection reagent (ratio 3:1 PEI:DNA).
- Harvest: Collect viral supernatant at 48 and 72 hours post-transfection, concentrate via PEG-it virus precipitation solution.
- Transduce: Infect target cell line (e.g., HEK293, CHO) with viral particles in the presence of 8 µg/mL polybrene. Spinfect at 800 x g for 45 minutes at 32°C.
- Select: Apply appropriate antibiotic (e.g., 2 µg/mL puromycin) for 7 days to establish stable pool.
3. Directed Evolution Cycling & Variant Isolation Cycles of mutagenesis and selection drive evolution.
Protocol 2: Iterative Evolution Cycle using Doxycycline-Induced Mutation & FACS Objective: Conduct rounds of mutation and phenotypic selection to enrich for improved variants.
- Induce Mutagenesis: Add 1 µg/mL doxycycline to the stable cell pool to induce target gene expression and 1 µM of the relevant base editor activator (e.g., for A3G-BE, add 1 µM APOBEC1 activator) for 72 hours.
- Apply Selection Pressure: Subject the mutated population to the defined selective condition (e.g., add a cytotoxic drug if target is a detoxifying enzyme; culture at low temperature if target is a cold-sensitive enzyme) for 5-7 days.
- Sort/Isolate: For fluorescent or surface-displayed phenotypes, use Fluorescence-Activated Cell Sorting (FACS). Harvest cells, resuspend in PBS + 2% FBS, and sort the top 1-5% of the population based on signal intensity. Plate single cells into 96-well plates.
- Recover & Expand: Culture sorted single cells for 7-14 days.
- Characterize Clones: Screen clonal populations via functional assay. Harvest genomic DNA from top performers using a commercial kit.
- Sequence: Amplify target gene locus from gDNA and perform Sanger or NGS to identify mutations.
Table 2: Evolution Cycle Quantitative Benchmarks
| Cycle Stage | Typical Duration | Success Metric |
|---|---|---|
| Mutagenesis Phase | 3-5 days | >30% cell viability post-induction |
| Selection Phase | 5-10 days | 10-100x enrichment of population signal |
| Single-Cell Sorting | 1 day | >50% clonal outgrowth rate |
| Clone Screening | 7-14 days | Identification of >3 clones with >2x improved activity |
4. Final Variant Validation Isolated variants require orthogonal validation.
- Re-cloning & Recombinant Expression: Subclone the mutant ORF into an expression vector, purify the protein.
- Biophysical Characterization: Determine kinetic parameters (Km, kcat), thermostability (Tm by DSF), and binding affinity (KD by SPR/BLI).
- In Vivo/Functional Validation: Test in the final application context (e.g., animal model, production bioreactor).
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent/Material | Function & Rationale |
|---|---|
| Lenti-X Bx Packaging System (Takara) | High-titer, 3rd generation lentiviral packaging plasmids for safe, efficient stable cell line generation. |
| PEI MAX 40K (Polysciences) | High-efficiency, low-toxicity transfection reagent for plasmid delivery in packaging cells. |
| HyClone Fetal Bovine Serum (Cytiva) | Consistent, high-performance serum for cell culture during critical selection and outgrowth phases. |
| CloneR Supplement (STEMCELL) | Enhances single-cell survival and clonal outgrowth post-FACS, crucial for monoculture establishment. |
| KAPA HiFi HotStart ReadyMix (Roche) | High-fidelity PCR master mix for accurate amplification of target loci from genomic DNA for sequencing. |
| SNAPgene Software | Essential for molecular biology design, visualization, and precise planning of genetic constructs. |
| Anti-Cas9 Antibody (7A9-3A3, Cell Signaling) | Validates Cas9 protein expression in engineered cell lines via Western blot. |
| NucleoSpin Tissue Kit (Macherey-Nagel) | Reliable gDNA isolation from mammalian cells for subsequent PCR and sequence analysis of evolved variants. |
Directed Evolution Workflow Overview
CRISPR-Cas Mutagenesis Pathways
Within the broader thesis on CRISPR-Cas mediated directed evolution, the design of specialized CRISPR libraries is foundational. These libraries enable the systematic perturbation of genomes to engineer proteins, pathways, and cellular functions. This Application Note details three core library design strategies—Saturation Mutagenesis, Domain Targeting, and Random Insertion—providing protocols and resources for their implementation in drug discovery and functional genomics.
Library Design Strategies: Comparative Analysis
Table 1: Key Characteristics of CRISPR Library Strategies
| Strategy | Primary Goal | Typical Library Size | Key Cas Enzyme | Editing Outcome | Primary Application in Directed Evolution |
|---|---|---|---|---|---|
| Saturation Mutagenesis | Interrogate all possible amino acid substitutions at defined residues. | 10^2 - 10^4 variants per target | Cas9-nickase (nCas9) fused to deaminase (e.g., BE), or Cas9-DD (Diversity Descriptor) | Targeted point mutations. | Protein affinity maturation, stability engineering. |
| Domain Targeting | Disrupt, delete, or swap specific protein functional domains. | 10^2 - 10^3 variants | Cas9 (cleavage), CRISPR/Cas-derived recombinases (e.g., Cas9-RecT). | Large deletions, domain replacements. | Elucidating domain function, creating chimeric proteins. |
| Random Insertion | Integrate diverse sequences (e.g., tags, peptides, coding exons) randomly into the genome. | 10^5 - 10^7 variants | Cas9 fused to transposase (e.g., Cas9-Tn7), or CRISPR-associated recombinase. | Precise sequence insertion. | Functional domain scanning, reporter integration, gain-of-function screens. |
Table 2: Quantitative Comparison of Delivery and Efficiency
| Parameter | Saturation Mutagenesis (Base Editing) | Domain Targeting (Dual sgRNA) | Random Insertion (CRISPR-Associated Transposition) |
|---|---|---|---|
| Indel Efficiency Range | N/A (Not double-strand break dependent) | 20-40% (for deletion formation) | N/A (Insertion is precise) |
| HDR/Insertion Efficiency | 10-50% (Base conversion) | Low (<10% for HDR-based replacement) | 10-30% (In E. coli); 1-10% (In mammalian cells) |
| Typical Delivery Method | Lentiviral vector | Plasmid or RNP transfection | Plasmid transfection (often requires donor plasmid) |
| Off-target Potential | Moderate (Guide-dependent) | High (Two guides increase risk) | Low (Transposase integration has bias but is not guide-dependent) |
| Optimal Library Screening Format | FACS, phenotypic selection | PCR genotyping, antibiotic selection | Next-generation sequencing, phenotypic selection |
Detailed Experimental Protocols
Protocol 1: Saturation Mutagenesis via CRISPR-Cas9 Base Editing
Objective: To generate all possible single-nucleotide variants within a target codon window. Materials: See "Research Reagent Solutions" (Section 6). Procedure:
- Design sgRNAs: Design a single-guide RNA (sgRNA) targeting the genomic region of interest. The protospacer should position the target adenine (for ABE) or cytosine (for CBE) within the editing window (typically positions 4-8 for ABE8e, 3-10 for BE4max) of the base editor.
- Library Synthesis: Synthesize an oligo pool containing the sgRNA sequence with a randomized NN(N) sequence at the target codon site within the sgRNA scaffold region to create variant guides, OR clone the base editor and a fixed sgRNA and deliver with a pooled oligo donor library containing all possible codon substitutions.
- Delivery: Co-transfect HEK293T cells (or target cell line) with the following using a high-efficiency transfection reagent (e.g., Lipofectamine 3000):
- Plasmids encoding the base editor (e.g., BE4max-pCMV).
- Plasmid encoding the sgRNA library (if using variant guides) or a single sgRNA.
- (If using donor oligos) Pooled single-stranded oligodeoxynucleotide (ssODN) donor library.
- Harvest and Analysis: Harvest genomic DNA 72 hours post-transfection. Amplify the target region by PCR and submit for next-generation sequencing (NGS). Analyze results using tools like CRISPResso2 or BE-Analyzer to quantify editing efficiency and variant distribution.
Protocol 2: Domain Deletion via Dual sgRNA/Cas9 Cleavage
Objective: To create precise deletions of a specific protein domain encoded by exons 3-5. Procedure:
- Design Dual sgRNAs: Design two sgRNAs with high predicted efficiency, targeting the 5' and 3' boundaries of the genomic region (e.g., exon 3 start and exon 5 end). Ensure they are in the same orientation.
- Clone sgRNA Expression Constructs: Clone expression cassettes for both sgRNAs into a single vector (e.g., pX330 derivative with dual U6 promoters) or use two separate plasmids.
- Transfection and Deletion: Co-transfect the target cell line with the dual sgRNA plasmid(s) and a plasmid expressing SpCas9 (if not expressed from the sgRNA vector). A non-homologous end joining (NHEJ) repair pathway will ligate the distal ends, excising the intervening sequence.
- Screening: 5-7 days post-transfection, isolate genomic DNA. Perform PCR with primers flanking the deletion target. Successful deletion will yield a smaller product. Confirm by Sanger sequencing.
Protocol 3: Random Peptide Insertion via CRISPR-Associated Transposase (CAST)
Objective: To randomly integrate a defined peptide tag sequence across the genome for gain-of-function screening. Procedure:
- Assemble CAST Components: The CAST system requires three plasmids for mammalian delivery: a) Transposase fused to catalytically dead Cas9 (dCas9), b) Transposon donor plasmid containing the peptide tag sequence flanked by transposon ends, c) Plasmid expressing the sgRNA targeting a specific genomic locus (for targeted random insertion within a window) or a library of sgRNAs.
- Library Transfection: Co-transfect the three plasmids at an optimized molar ratio (e.g., 1:1:1) into the target cell population.
- Selection and Expansion: Apply appropriate antibiotic selection 48 hours post-transfection to select for cells that have integrated the transposon (which contains a resistance marker). Expand the population for 7-10 days.
- Phenotypic Screening & Analysis: Perform the relevant phenotypic screen (e.g., drug resistance, FACS for a surface marker). Recover genomic DNA from selected and control populations. Use sequencing of the transposon-genome junctions (e.g., using linear amplification-mediated PCR - LAM-PCR) to identify integration sites.
Visualization of Workflows and Relationships
Diagram 1: Saturation Mutagenesis via Base Editing Workflow
Diagram 2: Domain Targeting via Dual sgRNA Deletion
Diagram 3: Random Insertion via CRISPR-Associated Transposase
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR Library Construction and Screening
| Reagent / Solution | Function / Description | Example Product / Kit |
|---|---|---|
| High-Fidelity DNA Polymerase | For accurate amplification of library components and target sequences for NGS. | Q5 High-Fidelity DNA Polymerase (NEB). |
| Pooled sgRNA Library Oligos | Synthesized oligonucleotide pool encoding the variant sgRNA sequences. | Custom oligo pool synthesis (Twist Biosciences, IDT). |
| Base Editor Plasmid | Mammalian expression vector for cytosine (CBE) or adenine (ABE) base editor. | pCMV-BE4max (Addgene #112093), pCMV-ABE8e (Addgene #138495). |
| Lentiviral Packaging Mix | For generating high-titer lentiviral particles to deliver libraries to hard-to-transfect cells. | Lenti-X Packaging Single Shots (Takara Bio). |
| Next-Generation Sequencing Service | For deep sequencing of edited pools to quantify variant abundance. | Illumina NovaSeq 6000, MiSeq. |
| CRISPR-Cas9 Transposase System | All-in-one or modular plasmids for CAST. | pCAST (Mosaic-like) system (e.g., pCAST-hyPBase from Addgene #103922). |
| Genomic DNA Extraction Kit | For high-quality, PCR-ready gDNA from cultured mammalian cells. | DNeasy Blood & Tissue Kit (Qiagen). |
| Cell Line with High HDR/NHEJ Efficiency | Engineered cell line for optimal CRISPR editing outcomes. | HEK293T, HAP1, or cell lines expressing Cas9 (e.g., HEK293T-3xFlag-Cas9). |
| NGS Analysis Software | Bioinformatics tool for quantifying editing outcomes and variant frequencies from sequencing data. | CRISPResso2, MAGeCK, pinAPL-Py. |
Application Notes
Within a CRISPR-Cas mediated directed evolution framework, the efficient delivery and stable integration of diverse genetic libraries into host cells is a critical first step. The choice of method directly impacts library complexity, uniformity, and the subsequent fitness screen's validity. Key considerations include payload size, host cell type (mammalian, bacterial, yeast), desired integration profile (random vs. targeted), and transformation/transfection efficiency.
Recent advances have moved beyond single-vector systems to hybrid strategies combining high-capacity delivery with high-efficiency, Cas-mediated targeted integration. This enables the introduction of vast variant libraries (10^8-10^10 members) into specific genomic safe harbors, minimizing positional effects and enabling comparative functional assays.
Quantitative performance metrics for common methods are summarized below:
Table 1: Comparative Analysis of Library Delivery & Integration Methods
| Method | Max Payload (approx.) | Typical Efficiency (Mammalian) | Integration Type | Key Advantage | Key Limitation |
|---|---|---|---|---|---|
| Lentiviral Transduction | ~8 kb | High (≥80% transducibility) | Random, stable | Broad tropism; stable expression in dividing/non-dividing cells | Size constraint; biosafety level 2+ |
| AAV Transduction | ~4.7 kb | Moderate to High | Primarily episomal | Low immunogenicity; high titer possible | Very small payload; complex production for library scales |
| Electroporation (plasmid) | >10 kb | Variable (5-60%) | Random, stable (if contains ITR/transposon) | Simplicity; large payload | High cell mortality; requires optimized protocols per cell type |
| Lipid Nanoparticles (mRNA) | N/A (encodes Cas9/gRNA) | High (≥70% protein expression) | Enables HDR (co-delivery with donor) | Low toxicity; high efficiency in hard-to-transfect cells | Transient Cas9 expression; donor template requires separate delivery |
| Nucleofection (RNP + donor) | Donor dependent | Moderate (20-40% HDR) | Targeted (HDR) | Rapid, precise; reduces off-target integration | Throughput can be lower; optimized kits per cell line |
| VLP-mediated Delivery | ~5 kb (for Cas9/gRNA) | Moderate (10-30% editing) | Targeted (as RNP) | Non-viral, transient; avoids plasmid integration | Lower efficiency than viral methods; nascent technology |
| Bacterial Conjugation | >100 kb | High (for prokaryotes/yeast) | Random or targeted (with engineered systems) | Extremely large payloads (e.g., whole pathway libraries) | Primarily for prokaryotes and some fungi |
Detailed Protocols
Protocol 1: Lentiviral Library Production and Transduction for Mammalian Cell Pools
Objective: Generate a pooled mammalian cell population with stably integrated variant libraries. Materials: Packaging plasmids (psPAX2, pMD2.G), transfer plasmid with library, 293FT cells, PEI transfection reagent, Polybrene (8 µg/mL), PBS, serum-containing medium, 0.45 µm filter, ultracentrifuge.
- Library Virus Production: Seed 293FT cells in 15-cm dishes to reach 70-80% confluency at transfection. For each dish, prepare a transfection mix in Opti-MEM: 20 µg library transfer plasmid, 15 µg psPAX2, 10 µg pMD2.G, and 90 µL PEI. Incubate 20 min, add dropwise to cells. Replace medium after 6-8 hours.
- Harvest and Concentrate: Collect viral supernatant at 48 and 72 hours post-transfection. Pool, filter through a 0.45 µm filter. Concentrate via ultracentrifugation (25,000 rpm, 2h, 4°C). Resuspend pellet in cold PBS, aliquot, and titre on target cells.
- Transduction for Library Generation: Seed target cells (e.g., HEK293T, HeLa) at 25% confluency. The next day, add viral supernatant at a low MOI (Multiplicity of Infection) of ~0.3-0.4 with Polybrene to ensure most cells receive a single integration. Spinfect at 1000 x g for 90 min at 32°C. Return to incubator.
- Selection and Expansion: 48 hours post-transduction, add appropriate selection antibiotic (e.g., Puromycin). Maintain selection for at least 5-7 days until all non-transduced control cells are dead. Expand the polyclonal pool for downstream screening.
Protocol 2: CRISPR-HDR Mediated Targeted Integration via Nucleofection of RNP and dsDNA Donor
Objective: Integrate a variant library into a defined genomic locus via homology-directed repair (HDR) in mammalian cells. Materials: Cas9 nuclease (protein), sgRNA (targeting genomic safe harbor), dsDNA donor template with homology arms (≥400 bp) and library, Amaxa Nucleofector and appropriate kit (e.g., SF Cell Line Kit), pre-warmed medium.
- RNP Complex Formation: For one reaction, mix 10 pmol of purified Cas9 protein with 30 pmol of synthetic sgRNA in Nucleofector solution. Incubate at room temperature for 10-20 minutes.
- Cell Preparation: Harvest and count target cells. Centrifuge and resuspend in PBS. For each nucleofection, use 1-2 x 10^5 cells.
- Nucleofection: Combine cells, RNP complex, and 200-500 ng of dsDNA donor library template in a Nucleofector cuvette. Select the appropriate pre-optimized program (e.g., FF-113 for HEK293). After nucleofection, immediately add pre-warmed medium and transfer to a culture plate.
- Recovery and Analysis: Culture cells for 48-72 hours. Allow recovery before any selection. Analyze integration efficiency via genomic PCR and NGS of the target locus across the pool. Apply selection if the donor contains a resistance marker.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Library Delivery and Integration
| Item | Function in Context |
|---|---|
| Lenti-X Concentrator | Simplifies lentivirus concentration via precipitation, avoiding ultracentrifugation. |
| TransIT-293 Transfection Reagent | High-efficiency, low-toxicity reagent for plasmid delivery into packaging cell lines. |
| Alt-R S.p. Cas9 Nuclease V3 | High-activity, recombinant Cas9 protein for RNP formation, ensuring rapid, transient activity. |
| CleanCap Cas9 mRNA | Co-transfection-ready, 5' capped and polyadenylated mRNA for transient, high-level Cas9 expression. |
| Neon Transfection System | Electroporation-based platform for high-efficiency delivery of RNPs/donor DNA into difficult cell types. |
| Gibson Assembly Master Mix | Enables seamless, one-pot assembly of large donor DNA fragments with homology arms and library inserts. |
| ClonePlus Screen | Enhances viability of difficult-to-transfect cells post-electroporation/nucleofection, improving yield. |
| Next-Gen Sequencing Kits (e.g., Illumina MiSeq) | Essential for assessing library representation pre- and post-integration, and after screening. |
Visualizations
CRISPR-Cas systems have revolutionized functional genomics and directed evolution. A critical component of CRISPR-mediated directed evolution is the integration of efficient selection and screening platforms to isolate rare variants with desired phenotypes. This document details current application notes and protocols for three primary platforms—Fluorescence-Activated Cell Sorting (FACS), survival assays, and reporter systems—within the context of accelerating directed protein evolution and functional genomics research.
FACS-Based Enrichment for CRISPR-Modified Cells
Application Notes
Fluorescence-Activated Cell Sorting (FACS) is a powerful, high-throughput method to isolate cells based on fluorescence signals linked to CRISPR editing outcomes. It is particularly valuable for directed evolution to select variants with enhanced binding, enzymatic activity, or expression levels. Recent advances integrate CRISPR barcoding with FACS to track lineage and phenotype simultaneously.
Protocol: FACS for CRISPR-Mediated Surface Display Evolution
Objective: To isolate yeast or mammalian cells displaying a protein variant of interest with enhanced binding properties from a CRISPR-mutagenized library.
Key Research Reagent Solutions:
| Reagent/Material | Function |
|---|---|
| CRISPR-Cas9 Ribonucleoprotein (RNP) Complex | Directs targeted double-strand breaks to the gene of interest. |
| Homology-Directed Repair (HDR) Template Library | A pool of oligonucleotides containing diverse mutations for HDR. |
| Fluorescently-Conjugated Ligand/Antibody | Binds to the displayed protein, providing a fluorescence signal for sorting. |
| Cell Strain Optimized for Surface Display (e.g., Yeast, HEK293) | Host for protein display and CRISPR editing. |
| NGS Library Prep Kit | For validating sorted pool diversity and enrichment. |
Procedure:
- Library Generation: Co-electroporate your chosen cell line with CRISPR-Cas9 RNP complex and the ssDNA HDR template library targeting the surface display gene.
- Recovery: Allow cells to recover for 48-72 hours in appropriate media to enable editing and expression of variants.
- Staining: Harvest cells, wash with PBS + 1% BSA, and incubate with the fluorescent ligand or antibody at a predetermined, sub-saturating concentration (e.g., 10-100 nM) for 30 min on ice.
- Washing: Wash cells twice to remove unbound label.
- FACS Sorting: Resuspend cells in sorting buffer. Use a high-performance sorter (e.g., Sony SH800, BD FACSAria). Gate on live, single cells, then collect the top 0.5-5% of cells with the highest fluorescence intensity. Perform a "no-stain" control to set the negative gate.
- Expansion & Iteration: Culture sorted cells to expand the population. Repeat the sorting process for 2-4 additional rounds to enrich for high-binders.
- Analysis: Sequence the target region from the final pool and individual clones to identify enriched mutations.
Survival/Selection Assays
Application Notes
Survival assays apply a direct selective pressure (e.g., antibiotic resistance, nutrient auxotrophy, toxic compound) where only cells with a specific CRISPR-induced edit can proliferate. This positive-negative selection is a cornerstone for gene essentiality studies (CRISPR knockout screens) and for evolving enzymes with new functions under lethal conditions.
Protocol: CRISPR-Cas9 Mediated Directed Evolution of an Antibiotic Resistance Enzyme
Objective: To evolve a β-lactamase variant with activity against a novel β-lactam antibiotic using a survival-based selection.
Key Research Reagent Solutions:
| Reagent/Material | Function |
|---|---|
| M9 Minimal Media Plates | Provides defined medium for selection. |
| Novel β-lactam Antibiotic (e.g., Cefotaxime) | Selective pressure; only cells with active evolved enzyme survive. |
| Lentiviral CRISPR Library (e.g., Brunello) | For genome-wide knockout screening in essential gene identification. |
| Error-Prone PCR Kit | To generate mutations in the target enzyme gene. |
| Plasmid expressing dCas9-Fused Transcriptional Activator (CRISPRa) | To upregulate the mutant enzyme library for selection. |
Procedure:
- Mutant Library Creation: Use error-prone PCR on the β-lactamase gene and clone it into an inducible expression plasmid.
- CRISPR-Mediated Activation: Co-transform E. coli with the mutant plasmid library and a CRISPRa system (dCas9-activator) targeting the promoter of the chromosomal copy of a native, non-essential gene (as a proof-of-concept).
- Selection: Plate the transformed library onto M9 agar plates containing a high concentration of the novel β-lactam antibiotic (e.g., 100 µg/mL cefotaxime). Include a control plate with ampicillin to ensure baseline function.
- Incubation: Incubate at 37°C for 24-48 hours. Only colonies expressing a β-lactamase variant capable of hydrolyzing the drug will grow.
- Harvest & Validation: Pool surviving colonies, extract plasmid DNA, and retransform fresh cells to confirm phenotype. Sequence the gene from resistant clones.
- Iteration: Use the sequences from the first round of survivors as a template for a subsequent, more diversified library and repeat selection at higher antibiotic concentrations.
Table: Example Survival Data for β-lactamase Evolution
| Selection Round | Antibiotic Concentration (µg/mL) | Colonies Surviving | Library Diversity Pre-Selection (Unique Variants) |
|---|---|---|---|
| 1 | 50 | ~1,200 | 1.0 x 10^7 |
| 2 | 200 | ~350 | 5.0 x 10^5 |
| 3 | 500 | ~45 | 2.0 x 10^4 |
Reporter Systems
Application Notes
Reporter systems convert a desired molecular event (e.g., transcriptional activation, protein-protein interaction, enzymatic activity) into a quantifiable signal like fluorescence or luminescence. CRISPR-compatible reporters are essential for high-throughput screening of gRNA efficacy, regulatory element activity, and in directed evolution of transcriptional factors or biosynthetic pathways.
Protocol: Dual-Fluorescence Reporter for CRISPRi/efficiency and Off-Target Assessment
Objective: To simultaneously monitor CRISPR-mediated knockdown and a transfection/viability control using a dual-fluorescence reporter.
Key Research Reagent Solutions:
| Reagent/Material | Function |
|---|---|
| Dual-Reporter Plasmid (e.g., pmirGLO-based) | Contains Target (e.g., GFP) and Control (e.g., RFP) genes. |
| Lipofectamine CRISPRMAX | For efficient delivery of CRISPR RNP or plasmids. |
| dCas9-KRAB Repressor (CRISPRi) | For targeted transcriptional repression. |
| Flow Cytometer (not sorter) | For quantifying population-level fluorescence shifts. |
Procedure:
- Reporter Construction: Clone a gRNA target sequence specific to your gene of interest into the 3'UTR of the GFP gene on a dual-reporter plasmid. RFP is driven by a separate, constitutive promoter.
- Cell Transfection: Seed HEK293T cells in a 24-well plate. Co-transfect with:
- The dual-reporter plasmid (100 ng)
- Plasmid expressing dCas9-KRAB and the targeting gRNA (400 ng)
- Use Lipofectamine 3000 per manufacturer's protocol.
- Incubation: Incubate cells for 72 hours to allow for repression and reporter turnover.
- Analysis: Harvest cells, wash with PBS, and analyze on a flow cytometer. Measure median fluorescence intensity (MFI) for GFP and RFP channels.
- Data Normalization: For each cell, normalize GFP signal to RFP signal to control for transfection efficiency and cell size. Calculate the knockdown efficiency as: (Normalized GFP MFI with gRNA)/(Normalized GFP MFI with non-targeting gRNA) x 100%.
- Application in Directed Evolution: This system can be adapted to evolve dCas9-KRAB or gRNA variants for enhanced specificity by using the RFP/GFP ratio as a screen for on-target vs. off-target effects.
Integrated Platform Comparison & Data
Table: Comparison of CRISPR-Compatible Selection & Screening Platforms
| Platform | Throughput | Quantitative Output | Key Application in Directed Evolution | Typical Timeline (Excluding NGS) | Cost |
|---|---|---|---|---|---|
| FACS | Very High (10^7-10^8 cells) | Yes (Fluorescence Intensity) | Evolving binding affinity, catalytic activity (via substrates), expression levels. | 1-2 weeks per round | High (Equipment, reagents) |
| Survival Assay | High (10^8-10^10 cells) | No (Binary Live/Dead) | Evolving antibiotic/toxin resistance, metabolic pathway engineering, essential gene identification. | 1 week per round | Low to Medium |
| Reporter System (Microscopy/Flow) | High (10^5-10^7 cells) | Yes (Luminescence/Fluorescence) | Evolving transcriptional regulators, optimizing CRISPR tool efficiency, biosensor development. | 1-2 weeks per screen | Medium |
The integration of CRISPR-Cas systems into directed evolution platforms represents a paradigm shift in protein engineering. Within the broader thesis of CRISPR-Cas-mediated directed evolution, this approach transcends traditional random mutagenesis by enabling precise, trackable, and efficient diversification of genomic loci combined with powerful phenotypic selection. This application note details a methodology for leveraging this capability to engineer therapeutic antibodies with enhanced affinity and stability, two critical determinants of efficacy, manufacturability, and dosing.
Key Experimental Data and Outcomes
Table 1: Comparative Analysis of Antibody Engineering Platforms
| Platform Feature | CRISPR-Cas Directed Evolution | Error-Prone PCR/ Yeast Display | Site-Saturation Mutagenesis |
|---|---|---|---|
| Mutation Introduction | Targeted, genomic, combinatorial | Random, plasmid-based, in vitro | Targeted, limited to predefined sites |
| Library Diversity | Very High (10^7-10^9) | Moderate (10^7-10^8) | Low (≤ 400 per site) |
| Throughput Screening | FACS-based (10^8 cells) | FACS-based (10^8 cells) | Medium-throughput (ELISA/SPR) |
| Mutation Tracking | Integrated via NGS of genomic DNA | Plasmid sequencing | Individual clone analysis |
| Primary Application | Affinity, stability, & developability | Affinity maturation | Affinity optimization at hot spots |
| Typical KD Improvement | 10 - 1000-fold | 10 - 100-fold | 5 - 50-fold |
| Tm Increase Achieved | +5°C to +15°C | +2°C to +8°C | +3°C to +10°C |
Table 2: Exemplar Results from a CRISPR-Cas Antibody Maturation Campaign
| Antibody Clone | Target Antigen | Wild-Type KD (nM) | Evolved KD (nM) | Fold Improvement | Tm (°C) | Aggregation Propensity |
|---|---|---|---|---|---|---|
| WT-1A2 | IL-6R | 4.5 | 0.045 | 100x | 67 | Moderate |
| EV-1A2.1 | IL-6R | 4.5 | 0.018 | 250x | 72 | Low |
| EV-1A2.7 | IL-6R | 4.5 | 0.032 | 140x | 74 | Very Low |
| WT-3B4 | TNF-α | 12.1 | 0.21 | 58x | 63 | High |
| EV-3B4.3 | TNF-α | 12.1 | 0.11 | 110x | 68 | Moderate |
Detailed Protocol: CRISPR-Cas Mediated Library Generation & Selection in Mammalian Cells
Phase 1: sgRNA Design & Donor Library Construction
- Target Identification: Identify antibody gene regions for diversification (e.g., Complementarity-Determining Regions (CDRs), framework regions affecting stability).
- sgRNA Design: Design 2-4 sgRNAs flanking the target region. Cloning: Clone sgRNAs into a lentiviral vector (e.g., lentiCRISPRv2).
- Oligo Library Synthesis: Design a degenerate oligonucleotide pool encoding targeted mutations. Use trinucleotide codons to minimize amino acid bias. Include homology arms (≥ 35 bp) matching sequences upstream/downstream of the CRISPR cut site.
- Donor Library Preparation: Amplify the oligo pool via PCR. Purify using a size-selection gel or beads.
Phase 2: Library Delivery & Integration
- Cell Line Preparation: Culture a mammalian cell line (e.g., CHO, HEK293) stably expressing the parental antibody. Ensure high viability (>95%).
- Co-transfection: Co-transfect cells with:
- sgRNA/Cas9 expression plasmid (1 µg)
- ssDNA or dsDNA donor library (3 µg) using a high-efficiency transfection reagent (e.g., PEI MAX).
- Recovery & Selection: Allow recovery for 48 hours. Apply selection (e.g., puromycin for sgRNA vector) for 5-7 days to enrich for successfully transfected/infected cells.
Phase 3: FACS-Based Screening for Affinity & Stability
- Antigen Labeling: Label target antigen with distinct fluorophores (e.g., AF488, PE).
- Stability Probe: Use a fluorescent dye (e.g., SYPRO Orange) that binds to exposed hydrophobic patches of denatured protein.
- Stability Challenge: Aliquot cells and incubate at varying temperatures (e.g., 60°C, 65°C, 70°C) for 10 minutes to partially denature unstable variants.
- Multi-Parameter FACS Sort: Perform sorting on live, single cells:
- Gate 1: High antigen-binding signal (AF488++, PE++) at 37°C (high affinity).
- Gate 2: Low SYPRO Orange signal (low dye binding) post-thermal challenge (high stability). Collect top 0.5-1% of the population meeting both criteria.
- Recovery & Expansion: Sort cells directly into 96-well plates or culture media. Expand for 7-14 days.
Phase 4: Clone Analysis & Validation
- Supernatant Screening: Measure antibody titer (ELISA) and binding affinity (via flow cytometry or Octet BLI) from supernatant.
- Genomic DNA Extraction & NGS: Isolate gDNA from top clones. PCR-amplify the targeted antibody region and submit for Next-Generation Sequencing (NGS) to identify mutations.
- Deep Mutational Scanning Analysis: For pooled libraries, extract gDNA from pre-sort and post-sort populations. Perform NGS and analyze enrichment/depletion of variants to map fitness landscapes.
- Reformat & Characterize: Clone V-genes from lead candidates into IgG expression vectors. Express and purify antibodies for definitive characterization (SPR/BLI for kinetics, DSC/DSF for Tm, SEC-MALS for aggregation).
Visualizations
Title: CRISPR-Cas Antibody Engineering Workflow
Title: Dual-Parameter FACS Screening Strategy
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for CRISPR-Cas Antibody Directed Evolution
| Reagent/Material | Function/Description | Example Vendor/Product |
|---|---|---|
| Lentiviral CRISPR Vector | Delivers sgRNA and Cas9 (e.g., SpCas9) for stable genomic integration and editing. | Addgene: lentiCRISPRv2, Takara Bio: pGuide-His |
| Custom Oligo Library | Degenerate nucleotide pool serving as the donor template for HDR, encoding the variant library. | Twist Bioscience, IDT (Trimer codon blocks) |
| High-Efficiency Transfection Reagent | Enables co-delivery of large DNA constructs (Cas9/sgRNA + donor library) into mammalian cells. | Polyethylenimine (PEI MAX), Lipofectamine 3000 |
| Fluorophore-Labeled Antigens | Critical for detecting antigen binding on the cell surface during FACS screening. | Bio-Techne, Thermo Fisher (Labeling kits) |
| Hydrophobic Dye (SYPRO Orange) | Binds to exposed hydrophobic regions of unfolded proteins; used as a stability sensor. | Thermo Fisher (S6650), Sigma-Aldrich |
| Cell Sorter (FACS) | Instrument for high-throughput, multi-parameter sorting based on fluorescence. | BD FACSAria, Beckman Coulter MoFlo Astrios |
| NGS Library Prep Kit | Prepares amplicons from genomic DNA for deep sequencing to identify enriched mutations. | Illumina Nextera XT, Swift Biosciences Accel-NGS |
| BLI/SPR Instrument | Label-free kinetic analysis of antibody-antigen interactions for definitive affinity measurement. | Sartorius Octet, Cytiva Biacore |
| Differential Scanning Calorimeter (DSC) | Gold-standard for measuring thermal unfolding midpoint (Tm) of purified antibodies. | Malvern MicroCal PEAQ-DSC |
Within the thesis on CRISPR-Cas mediated directed evolution, this application note details the integration of CRISPR tools to accelerate the engineering of enzymes for industrial biocatalysis and the discovery of novel functions. By enabling precise, multiplexed genome editing and efficient library generation, CRISPR-Cas systems move beyond traditional random mutagenesis, allowing for the targeted exploration of sequence-function relationships in enzyme-coding genes.
Key Quantitative Data
Table 1: Comparison of Directed Evolution Platforms for Enzyme Optimization
| Platform / Method | Mutation Rate (avg. per gene) | Library Size (typical) | Screening Throughput | Key Advantage | Primary Limitation |
|---|---|---|---|---|---|
| Error-Prone PCR (Traditional) | 1-10 mutations | 10⁴ - 10⁶ | 10³ - 10⁴ variants/day | Simplicity, broad mutation spectrum | Low frequency of beneficial mutations, laborious cycles |
| CRISPR-Cas9 Assisted MAGE | 1-5 precise mutations | 10⁸ - 10¹⁰ | N/A (selection-based) | High efficiency & precision in E. coli | Limited to tractable hosts, requires ssDNA design |
| CRISPR-BEST (Base Editing) | Single nucleotide variant (SNV) | 10⁷ - 10⁹ | 10⁵ - 10⁷ via selection | Direct C•G to T•A or A•T to G•C transitions without DSBs | Restricted to specific base changes, potential off-target edits |
| CRISPRi/dCas9 Screening | Gene expression modulation (knockdown) | Genome-wide (all genes) | 10⁸ - 10⁹ via NGS | Identifies optimal expression levels for pathway enzymes | Does not alter protein sequence directly |
Table 2: Performance Metrics of CRISPR-Optimized Industrial Enzymes (Recent Case Studies)
| Enzyme Class | Target Property | CRISPR Method Used | Rounds of Evolution | Improvement Fold | Application |
|---|---|---|---|---|---|
| PETase (polyester hydrolase) | Thermostability (Tm) | CRISPR-Cas9 with donor library (site-saturation) | 2 | Tm increase: +15°C | Plastic depolymerization |
| Transaminase (ATA-117) | Organic Solvent Tolerance | CRISPR-assisted multiplex automated genome engineering (MAGE) | 1 | Activity in 50% DMSO: 25x higher | Chiral amine synthesis |
| Cytochrome P450 (P450BM3) | Activity on Non-Native Substrate | dCas9-guided mutagenesis (targeted random) | 3 | Turnover number: 100x higher | Drug metabolite production |
| Lipase (CALB) | Enantioselectivity (E value) | Base Editor (CRISPR-BEST) | 1 | E from 12 to >200 | Pharmaceutical intermediate resolution |
Experimental Protocols
Protocol 1: CRISPR-Cas9 Mediated Saturation Mutagenesis of Enzyme Active Site
Objective: Generate a comprehensive library of single amino acid variants at a defined active site residue.
Materials: See "Research Reagent Solutions" below.
Procedure:
- Design & Synthesis: Design a degenerate oligonucleotide pool encoding all 20 amino acids at the target codon(s). Flank with 40-nt homology arms complementary to the genomic region. Clone into a donor plasmid.
- Plasmid Assembly: Co-transform the target microbial host (e.g., S. cerevisiae or B. subtilis) with two plasmids: (a) the donor plasmid, and (b) a CRISPR-Cas9 plasmid expressing a guide RNA (gRNA) targeting the wild-type sequence at the site.
- Induction & Editing: Induce Cas9 expression to create a double-strand break (DSB). The cell's homology-directed repair (HDR) machinery uses the donor library for repair, incorporating the mutations.
- Library Recovery & Screening: Harvest cells after 24-48h. Isolate genomic DNA and amplify the mutated gene region. Clone into an expression vector for high-throughput screening (e.g., microfluidic droplets, FACS) for the desired activity.
- Deep Sequencing Validation: Sequence the variant library pre- and post-selection via NGS to identify enriched mutations.
Protocol 2: CRISPR-dCas9 Assisted Continuous Evolution (CRISPR-ACE)
Objective: Evolve enzyme properties under a selective pressure in a turbidostat or chemostat setup.
Procedure:
- Strain Engineering: Integrate a dCas9-effector (e.g., transcriptional activator) system and a mutagenesis plasmid (e.g., expressing error-prone DNA polymerase) into the host genome.
- gRNA Library Design: Design a library of gRNAs targeting the promoter or coding region of the target enzyme gene. Clone into an inducible plasmid.
- Continuous Evolution: Dilute the culture continuously in a bioreactor under defined selective pressure (e.g., presence of toxic substrate, limiting carbon source). Periodically induce the mutagenesis system and the gRNA library.
- Monitoring & Sampling: Monitor population growth or product formation. Sample the culture at intervals over 100-500 generations.
- Variant Identification: Isolate genomic DNA from endpoint culture. Amplify and sequence the target gene from the population. Isolate single clones for characterization of improved variants.
Visualizations
Title: CRISPR Enzyme Engineering Workflow
Title: CRISPR-dCas9 for Tunable Enzyme Expression
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Supplier Examples | Function in CRISPR Enzyme Evolution |
|---|---|---|
| LentiCRISPR v2 Plasmid | Addgene | All-in-one vector for mammalian cell expression of Cas9 and gRNA. Useful for engineering enzyme-producing mammalian cell lines. |
| Beacon Optofluidic Platform | Berkeley Lights | Enables digital screening of single cells/enzymes in nanoliter droplets for activity, stability, or binding. |
| Gibson Assembly Master Mix | NEB | Enables seamless, one-step assembly of multiple DNA fragments for rapid construction of donor and gRNA expression plasmids. |
| NEBuilder HiFi DNA Assembly Kit | NEB | Similar to Gibson Assembly, for high-fidelity, scarless cloning of homology fragments and vector backbones. |
| Alt-R S.p. Cas9 Nuclease 3NLS | IDT | High-purity, recombinant Cas9 protein for efficient ribonucleoprotein (RNP) delivery, minimizing off-target effects. |
| CRISPRa dCas9-VPR Synergistic Activation Mediator | Addgene | A potent transcriptional activation system for upregulating endogenous enzyme genes or pathway genes. |
| T7 Endonuclease I | NEB | Detects insertion/deletion mutations (indels) caused by NHEJ repair, useful for assessing CRISPR cutting efficiency. |
| TruSeq DNA PCR-Free Library Prep Kit | Illumina | Prepares high-quality genomic DNA libraries for next-generation sequencing (NGS) of evolved variant pools. |
| GeneMorph II Random Mutagenesis Kit | Agilent | Introduces random mutations via error-prone PCR to generate diverse donor libraries for HDR. |
| CellASIC ONIX2 Microfluidic System | MilliporeSigma | Precisely controls culture environment for continuous evolution experiments in chemostat-like micro-environments. |
Application Notes
Directed evolution using CRISPR-Cas systems has emerged as a transformative strategy for engineering next-generation molecular tools. By coupling Cas-mediated mutagenesis with high-throughput screening, researchers can rapidly optimize protein-based biosensors for enhanced sensitivity, dynamic range, and specificity, while simultaneously advancing optogenetic actuators with improved light sensitivity, spectral selectivity, and kinetic properties. This iterative evolution cycle is central to a broader thesis on CRISPR-Cas mediated directed evolution, demonstrating its power to solve complex functional optimization problems beyond simple gene editing.
Key advancements include the evolution of fluorescent biosensors for neurotransmitters like dopamine and glutamate with sub-second kinetics and nanomolar affinity, enabling real-time monitoring of neuronal communication. For optogenetics, directed evolution has produced novel channelrhodopsin variants (e.g., ChRmine) with unprecedented light sensitivity, allowing for non-invasive neuronal stimulation deep within brain tissue. The integration of base editors and prime editors into these pipelines allows for precise, tunable mutagenesis (e.g., A>G transitions) to fine-tune specific protein properties without double-strand breaks, accelerating the development of clinical-grade tools.
Table 1: Evolved Biosensor Performance Metrics
| Tool Name | Target | Key Evolved Property | Original Value | Evolved Value | Screening Method |
|---|---|---|---|---|---|
| dLight1.3 | Dopamine | Binding Affinity (Kd) | ~200 nM (parent) | 90 nM | FACS (Fluorescence) |
| GRABGlu | Glutamate | Signal-to-Noise Ratio | ~50% ΔF/F | 230% ΔF/F | Microplate Fluorescence |
| iGluSnFR3 | Glutamate | Kinetics (τoff) | ~200 ms | 1.3 ms | Flow Cytometry |
| cADDis | cAMP | Dynamic Range (ΔR/R) | 1.0 | 3.5 | Transcriptional Reporter |
Table 2: Evolved Optogenetic Tool Variants
| Tool Name | Class | Key Evolved Property | Parent | Evolved Variant | Application |
|---|---|---|---|---|---|
| ChRmine | Channelrhodopsin | Photosensitivity (EC50) | 4.5 mW/mm² | 0.045 mW/mm² | Deep Brain Stimulation |
| Chronos | Channelrhodopsin | Kinetics (τoff) | ~10 ms | ~4 ms | High-Frequency Stimulation |
| BiPOLES | Bistable Step-Function Opsin | Light Sensitivity | Low (Requires high irradiance) | High (Single photon) | Long-Term Potentiation Studies |
| Jaws | Inhibitory Opsin (eNpHR) | Action Spectrum Peak | ~590 nm | ~630 nm (Red-shifted) | Reduced Phototoxicity |
Experimental Protocols
Protocol 1: CRISPR-Cas Mediated Directed Evolution of a GPCR-Based Biosensor
Objective: Evolve a genetically encoded biosensor for improved ligand affinity and fluorescent response.
Materials: See "Research Reagent Solutions" table.
Method:
- Library Construction: Design sgRNAs targeting the ligand-binding domain of the biosensor GPCR. Co-transfect a mammalian cell line (e.g., HEK293T) with:
- A plasmid encoding a hyperactive cytidine deaminase base editor (e.g., BE4max).
- The sgRNA plasmid pool.
- A donor template containing the biosensor gene (e.g., GRAB sensor) and a puromycin resistance gene.
- Mutagenesis & Selection: Culture cells for 72 hours under puromycin selection to enrich successfully edited cells.
- High-Throughput Screening: Harvest cells and resuspend in assay buffer. For a glutamate sensor, screen by:
- Loading cells into a FACS sorter equipped with a plate dispenser.
- Applying a pulse of saturating glutamate (e.g., 100 µM) directly in the fluidics stream.
- Sorting the top 0.1-1% of cells showing the highest rate of fluorescence increase (ΔF/Δt) into 96-well plates.
- Recovery & Iteration: Grow sorted cells, recover plasmid DNA, and sequence the biosensor gene from pooled populations. Clone individual variants and characterize in vitro. Use hits as the parent for subsequent evolution rounds.
Protocol 2: Evolution of Red-Shifted Channelrhodopsins via Orthogonal Replication
Objective: Generate channelrhodopsin variants with a red-shifted action spectrum.
Method:
- Diversification: Create a mutagenesis library of a channelrhodopsin (e.g., Chrimson) via error-prone PCR focused on residues near the retinal chromophore.
- Yeast Display & Screening: Clone the library into a yeast display vector fused to a surface epitope tag (e.g., HA). Induce expression.
- Photocurrent Proxy Screening: Use a two-step labeling process:
- Incubate yeast with an anti-HA primary antibody (to quantify surface expression).
- Incubate with a red fluorescent secondary antibody and a viability dye.
- Illuminate yeast culture with 630 nm light while simultaneously monitoring fluorescence via flow cytometry.
- Sort the population that maintains the highest viability (indicating minimal proton pumping/photo-toxicity under red light) AND high surface expression. This serves as a proxy for efficient, red-shifted membrane localization.
- Functional Validation: Isolate plasmid DNA from sorted yeast, transform into mammalian neurons, and validate photocurrent properties via patch-clamp electrophysiology under 630 nm illumination.
Diagrams
Title: Directed Evolution Workflow for Molecular Tools
Title: Biosensor Mechanism & Screening Logic
The Scientist's Toolkit
Table 3: Research Reagent Solutions for CRISPR-Driven Tool Evolution
| Item | Function in Protocol | Example Product/Catalog |
|---|---|---|
| Base Editor Plasmid | Mediates targeted C>T (or A>G) mutations without DSBs for fine-tuning. | BE4max (Addgene #112093) |
| sgRNA Library Pool | Guides Cas-deaminase fusion to target gene regions for diversification. | Custom synthesized oligo pool. |
| Mammalian Expression Vector | Cloning and expression of biosensor/opsin library in host cells. | pCAG (for neurons), pcDNA3.1. |
| FACS Sorter with Plate Dispenser | High-speed isolation of cells based on real-time fluorescent response. | BD FACSAria Fusion, Sony SH800. |
| Yeast Display Vector | Surface display of opsin libraries for expression-coupled screening. | pYD1 (Thermo Fisher). |
| Turbofect or Lipofectamine 3000 | High-efficiency transfection reagent for library delivery. | Thermo Fisher Scientific. |
| Patch Clamp Electrophysiology Rig | Gold-standard validation of evolved opsin ion channel function. | Molecular Devices Axopatch. |
| Modular Microplate Reader | Quantifying biosensor dynamic range and kinetics in population assays. | Tecan Spark, BMG CLARIOstar. |
Overcoming Pitfalls: Expert Strategies for Efficiency and Success
Within CRISPR-Cas mediated directed evolution, the core goal is to simulate and accelerate natural selection on a molecular scale. A persistent challenge is the effective maintenance of genetic diversity within combinatorial libraries throughout iterative selection rounds. Bottlenecks occur when stochastic sampling or selective pressures cause a severe reduction in library complexity, leading to the loss of rare but potentially high-value variants. This application note details protocols and strategies to monitor and preserve library diversity, ensuring comprehensive exploration of sequence-function space.
Quantitative Metrics for Diversity Assessment
Effective management requires quantitative tracking. Key metrics are summarized below.
Table 1: Key Quantitative Metrics for Library Diversity Assessment
| Metric | Measurement Method | Target Range (Ideal) | Interpretation |
|---|---|---|---|
| Library Size (Complexity) | NGS Census, Colony Forming Units (CFU) | >10^8 unique variants pre-selection | Baseline diversity. |
| Post-Selection Retention | (Pre-selection diversity) / (Post-selection diversity) | >1% of initial library | Indicates selection stringency. Severe bottlenecks show <<0.1%. |
| Variant Evenness (Shannon Index, H') | Calculated from NGS read counts per variant. H' = -Σ(pi * ln(pi)) | H' > 8 for large libraries (>10^7) | High evenness: most variants at similar abundance. Low evenness: dominance by few clones. |
| Clone Dominance | Percentage of total reads from the Top 10 most abundant variants. | <10% pre-selection; may increase post-selection. | >50% indicates a severe bottleneck or extremely strong selection. |
| Coverage Depth | Average NGS reads per unique variant. | >50-100x for reliable detection. | Ensures rare variants are detectable above sequencing noise. |
Protocol: Iterative Directed Evolution with Diversity Monitoring
This protocol integrates diversity checks into a standard CRISPR-Cas mediated directed evolution cycle.
Materials & Reagents:
- Target gene cloned into a CRISPR-compatible plasmid (e.g., with homology arms for HDR).
- sgRNA library targeting the gene for diversification (e.g., targeting mutational hotspots).
- In vitro or in vivo assembled Cas9/sgRNA ribonucleoprotein (RNP) complexes.
- Oligonucleotide donor library (ssODN or dsDNA) encoding designed mutations.
- Competent cells (e.g., NEB 10-beta for high transformation efficiency).
- Recovery media (e.g., SOC Outgrowth Medium).
- Selection plates with appropriate antibiotic or reporter-based screening system.
- QIAprep Spin Miniprep Kit or similar for plasmid recovery.
- Primers for NGS library preparation of the target locus.
Procedure:
- Library Transformation & Baseline Census (Round 0):
- Electroporate the pooled sgRNA/donor library with Cas9 RNP into competent cells. Include a "no-RNP" control to assess background.
- Plate a serial dilution for CFU counting to determine Library Size.
- Inoculate a liquid culture from the remainder and incubate. Isolate pooled plasmid DNA.
- Prepare an NGS amplicon library from the target locus. Sequence to establish baseline diversity, evenness (H'), and clone dominance.
Selection Round:
- Transform the Round 0 pooled library into the selection host (if different) or apply the selective pressure (e.g., antibiotic gradient, FACS sorting, reporter assay).
- Collect surviving/output cells. Plate a fraction for CFU to determine Post-Selection Retention.
- Isolve genomic DNA or plasmid from the output pool.
Diversity Assessment Post-Selection:
- Prepare and sequence an NGS amplicon library from the output pool.
- Analyze sequencing data (see Table 1). Calculate the Shannon Index (H') and Clone Dominance for the output pool. Compare to Round 0.
Library Regeneration & Bottleneck Mitigation:
- If metrics indicate excessive bottlenecking (e.g., H' drop >70%, Dominance >60%), implement mitigation:
- Backbone Dilution: If using a plasmid system, re-clone the enriched variant pool into fresh, naive backbone via Gibson Assembly or Golden Gate cloning to break linkage with potentially selected cis elements.
- Multiplexed Re-diversification: Use the enriched pool as template for error-prone PCR or additional oligo-directed mutagenesis focused on new regions before proceeding to the next selection round.
- Use the regenerated library as input for the next iterative selection round (return to Step 2).
- If metrics indicate excessive bottlenecking (e.g., H' drop >70%, Dominance >60%), implement mitigation:
Visualization
Diagram 1: Directed Evolution Workflow with Diversity Checkpoints
Diagram 2: Bottleneck Severity Decision Logic
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Diversity-Maintaining CRISPR Directed Evolution
| Reagent / Solution | Function & Importance for Diversity |
|---|---|
| High-Efficiency Electrocompetent Cells (e.g., NEB 10-beta, MegaX DH10B T1R) | Maximizes transformation efficiency (>10^9 CFU/µg) to ensure physical representation of large libraries, minimizing stochastic loss at the cloning step. |
| Pooled ssODN Donor Library | Provides the mutational template for HDR. Ultramer-quality pools ensure high-fidelity synthesis of complex, degenerate sequences, defining the library's theoretical diversity. |
| Next-Generation Sequencing (NGS) Kit (e.g., Illumina MiSeq Reagent Kit v3) | Enables deep, quantitative tracking of variant frequency and evenness across selection rounds. Essential for calculating diversity metrics. |
| CRISPR-Cas9 Nickase (Cas9n) or Base Editor | Reduces indels and improves HDR efficiency relative to wild-type Cas9, increasing the yield of precise, designed variants over a background of non-productive repair. |
| Magnetic Bead-Based Cleanup Kits (e.g., AMPure XP) | For consistent, high-recovery purification of pooled DNA libraries between PCR amplification and NGS steps, maintaining representative composition. |
| Gibson Assembly or Golden Gate Assembly Master Mix | For efficient library regeneration steps, allowing the transfer of enriched variant pools from a potentially biased plasmid backbone into a fresh genetic context. |
Application Notes
Within a thesis exploring CRISPR-Cas mediated directed evolution, a central and persistent challenge is the precise calibration of mutation induction with the maintenance of cellular viability and fitness. The objective is to generate sufficient genetic diversity for effective selection while preserving a functional cellular chassis for phenotype expression and screening. Excessive mutagenesis leads to synthetic lethality, overwhelming deleterious mutations, and population collapse. Insufficient mutagenesis fails to explore adaptive landscapes, stalling evolution.
Recent advancements (2023-2024) have focused on engineering temporal, spatial, and compositional control over CRISPR-based mutagenesis systems. Key strategies include:
- Tunable Editors: Using engineered or evolved Cas9 variants (e.g., "Cas9-OMe" with reduced activity) or modulating the expression levels of base editors (BEs) and prime editors (PEs) via inducible promoters or degradation tags.
- Combinatorial Targeting: Implementing multiplexed guide RNA (gRNA) libraries targeting specific genomic regions or pathways of interest, thereby concentrating diversity where it is most likely to yield beneficial phenotypes, sparing essential genes.
- Orthogonal Control Systems: Employing phage-derived non-homologous end joining (NHEJ) machinery (e.g., E. coli Pol I) or retroviral reverse transcriptases in conjunction with CRISPR targeting to decouple mutagenesis from canonical DNA damage response pathways.
- Continuous Evolution Platforms: Integrating mutagenesis modules with fluorescence-activated cell sorting (FACS) or continuous culture (e.g., eVOLVER) to dynamically adjust mutation rates based on real-time population fitness metrics.
Table 1: Performance Metrics of Selected Tunable Mutagenesis Systems
| System / Strategy | Typical Mutation Rate (per kb per gen.) | Cell Viability Impact (% of WT) | Key Regulatory Mechanism | Primary Use Case | Ref. (Year) |
|---|---|---|---|---|---|
| Hyperactive Cas9-nCas9 (D10A) | 10^-2 - 10^-1 | 15-40% | Constitutive expression; gRNA multiplexing | Global, high-diversity library generation | 2022 |
| T7 RNAP-Cas9 EvolvR | 10^-3 - 10^-2 | 60-80% | Polymerase processivity & promoter strength | Targeted, continuous evolution in bacteria | 2023 |
| Doxycycline-inducible AID-Cas9 | Tunable 10^-5 to 10^-3 | 70-95% (at low dose) | Tet-On promoter controlling AID expression | Mammalian cell directed evolution with temporal control | 2023 |
| Light-inducible Cas9-cytidine deaminase | Tunable 10^-6 to 10^-4 | >85% (at low irradiance) | Blue light-controlled dimerization | Spatiotemporally precise mutagenesis in biofilms | 2024 |
| CRISPR-X (dCas9-MS2-APOBEC1) | ~10^-3 at target loci | 70-90% | MS2 scaffold & APOBEC1 recruitment level | Localized mutagenesis around specific genomic sites | 2022 |
| OrthoRep (cytoplasmic T7/DdDp) | ~10^-5 per bp | >95% | Error-prone T7 RNAP/DdDp in cytoplasm | Continuous, orthogonal evolution in yeast | 2023 |
Table 2: Impact of Mutation Rate on Functional Clone Recovery in a Model Antibody Affinity Maturation Screen
| Induced Mutation Rate (per gene) | Library Size Screened | % Viable Cells Post-Mutagenesis | High-Affinity Hits Identified | % of Hits with Deleterious Off-Target Mutations |
|---|---|---|---|---|
| 0.001 | 1 x 10^8 | 92% | 3 | 10% |
| 0.01 | 1 x 10^8 | 65% | 12 | 25% |
| 0.1 | 1 x 10^8 | 22% | 1 | 80% |
| 1.0 | 1 x 10^8 | <5% | 0 | N/A |
Experimental Protocols
Protocol 1: Titrating Mutation Rate Using a Doxycycline-Inducible Base Editor in Mammalian Cells
Objective: To establish a dose-response relationship between inducer concentration, on-target mutation rate, and cell viability for calibrating directed evolution experiments.
Key Research Reagent Solutions:
| Reagent / Material | Function |
|---|---|
| HEK293T-Tet-On 3G Cells | Host cells with optimized, high-sensitivity doxycycline-inducible expression system. |
| Lenti-X Tet-On 3G Inducible Expression System | Lentiviral system for stable integration of the inducible BE construct. |
| pLVX-Tet3G-BE4max-P2A-mCherry | Plasmid encoding BE4max cytosine base editor and fluorescent reporter under TRE3GS promoter. |
| Target-specific sgRNA plasmid (e.g., pU6-sgRNA) | Drives BE to a specific genomic locus for mutation rate quantification. |
| Syncell Cell Viability Assay Kit | Fluorescent-based assay to distinguish live/dead cells without bias from edited phenotype. |
| Next-Generation Sequencing (NGS) Library Prep Kit for Amplicons | For precise quantification of editing efficiency and spectrum at target locus. |
Methodology:
- Stable Cell Line Generation: Co-transfect HEK293T-Tet-On 3G cells with the pLVX-Tet3G-BE4max and target sgRNA plasmid using a high-efficiency transfection reagent. Select with puromycin (2 µg/mL) for 7 days.
- Induction Gradient: Seed stable cells in a 12-well plate. At 50% confluency, treat with a doxycycline gradient (e.g., 0, 10, 50, 100, 500, 1000 ng/mL) in triplicate.
- Harvest and Analysis (72h post-induction):
- Viability: Detach cells from one set of wells. Stain with Syncell kit and analyze via flow cytometry. Calculate % viability relative to uninduced control.
- Genomic DNA Extraction: Harvest cells from parallel wells. Isolate gDNA using a silica-membrane column kit.
- Mutation Rate Quantification: Amplify the target locus from gDNA using high-fidelity PCR. Prepare NGS libraries and sequence on a MiSeq. Use CRISPResso2 or similar to calculate editing efficiency (% of reads with C•G to T•A conversions) and the distribution of mutations.
- Data Integration: Plot doxycycline concentration against both viability and editing efficiency. The optimal window for evolution experiments is the concentration yielding >60% viability and the desired mutation rate (e.g., 5-20%).
Protocol 2: Continuous Evolution in Yeast Using OrthoRep with Dynamic Mutation Rate Adjustment
Objective: To perform adaptive laboratory evolution under selective pressure while monitoring and adjusting the orthogonal mutagenesis rate to maintain population growth.
Key Research Reagent Solutions:
| Reagent / Material | Function |
|---|---|
| S. cerevisiae Strain with OrthoRep System | Engineered yeast where a cytoplasmic p1 plasmid is replicated by error-prone T7/DdDp polymerase. |
| Selection Plasmid (p1 derivative) | Plasmid housed in OrthoRep system, encoding the gene of interest (GOI) under selection. |
| Customized eVOLVER Hardware | Automated culturing device enabling real-time monitoring and adjustment of growth in multiple turbidostats. |
| Mutagenic Nucleotide Analogue (e.g., 5-Br-dUTP) | Can be fed to cells to further increase error rate of OrthoRep's polymerase. |
| qPCR Assay for p1 Plasmid Copy Number | Monitors plasmid stability under mutagenesis and selection. |
Methodology:
- System Setup: Clone the GOI into the p1 plasmid and transform into the OrthoRep yeast strain. Inoculate the strain into multiple eVOLVER vessels with complete synthetic media lacking a nutrient complemented by the GOI.
- Baseline Monitoring: Allow cultures to reach mid-log phase. Record baseline growth rates (OD600 via eVOLVER) and determine p1 copy number via qPCR.
- Induction and Dynamic Control: Initiate evolution by adding a low concentration of mutagenic analogue (e.g., 10 µM 5-Br-dUTP). Set eVOLVER to maintain OD600 within a defined range via dilution.
- Feedback Logic: Program a simple proportional control: If the growth rate (derived from OD600 over time) drops below 70% of the pre-induction baseline for 3 consecutive hours, automatically halve the concentration of mutagen in the media feed. If growth rate recovers and exceeds 90% for 6 hours, incrementally increase the mutagen concentration.
- Sampling and Analysis: Periodically sample cells for (a) GOI sequence analysis via NGS to track mutation accumulation, and (b) competitive fitness assays against the ancestral strain.
- Endpoint Cloning: After a predefined period (e.g., 100 generations), isolate the p1 plasmid from the population and from individual clones. Sequence the GOI and characterize improved variants.
Mandatory Visualizations
Title: Directed Evolution Workflow with Mutation Rate Optimization
Title: Trade-offs in Mutation Rate Tuning for Directed Evolution
Application Notes
Within CRISPR-Cas mediated directed evolution, precise and efficient genome editing is paramount for generating diverse mutant libraries and screening for desired phenotypes. The optimization of two core components—guide RNA (gRNA) design and Cas protein variant selection—directly dictates the success rate and outcome of evolutionary experiments. This document provides current protocols and data frameworks to empower researchers in these critical design phases.
1. Quantitative Comparison of Cas Variants for Directed Evolution The choice of Cas variant influences editing efficiency, precision, off-target profile, and PAM (Protospacer Adjacent Motif) flexibility. The following table summarizes key characteristics of contemporary Cas nucleases relevant to library generation.
Table 1: Comparison of Common CRISPR-Cas Variants for Genome Editing Applications
| Cas Variant | Native PAM | Size (aa) | Primary Editing Outcome | Key Advantage for Directed Evolution | Primary Limitation |
|---|---|---|---|---|---|
| SpCas9 | NGG | 1368 | DSB, NHEJ/HDR | High efficiency; well-characterized | Large size; restrictive PAM |
| SpCas9-NG | NG | ~1368 | DSB, NHEJ/HDR | Expanded targeting range (NG PAM) | Slightly reduced efficiency for some NG sites |
| xCas9(3.7) | NG, GAA, GAT | ~1368 | DSB, NHEJ/HDR | Broad PAM recognition | Inconsistency across cell types |
| SpRY | NRN > NYN | ~1368 | DSB, NHEJ/HDR | Near-PAMless targeting | Higher off-target potential |
| SaCas9 | NNGRRT | 1053 | DSB, NHEJ/HDR | Smaller size for viral delivery | Less flexible PAM |
| Cas12a (Cpfl) | TTTV | ~1300 | DSB, NHEJ/HDR (staggered cut) | Shorter crRNA; multiplexing from single transcript | Lower efficiency in some mammalian systems |
| Base Editors (BE) | Varies by Cas domain | ~1600 | Point Mutation (C•G to T•A or A•T to G•C) | Efficient, precise point mutation without DSBs; ideal for scanning mutagenesis | Limited to transition mutations; bystander editing |
| Prime Editors (PE) | Varies by Cas domain | ~2400 | Small Insertions, Deletions, all Base Subs | Versatile; templated edits without DSBs | Complex delivery; variable efficiency |
2. Guide RNA Design Parameters and Optimization For a given Cas variant, gRNA design is critical. Key parameters include on-target efficiency prediction and off-target minimization.
Table 2: Key Parameters for gRNA Design and Validation
| Parameter | Consideration | Tool/Measurement Method |
|---|---|---|
| On-Target Score | Predicts cleavage efficiency based on sequence features (e.g., GC content, nucleotide composition). | Rule Set 1, DeepCRISPR, Azimuth, ChopChop. |
| Off-Target Potential | Number and location of genomic sites with high sequence homology to the spacer. | Cas-OFFinder, GuideScan, MIT CRISPR Design Tool. |
| Seed Region | Bases 1-12 proximal to PAM are most critical for binding. Mismatches here often abolish cutting. | Ensure perfect homology in seed region for on-target. |
| Secondary Structure | gRNA or crRNA folding can impede Cas protein binding. | Check using RNAfold or internal algorithms in design tools. |
| Genomic Context | Target site chromatin accessibility (e.g., ATAC-seq data). | FAIRE-seq, DNase-seq data integration; consider using Cas9 derivatives with chromatin modulators. |
Experimental Protocols
Protocol 1: In Silico Design and Selection of gRNAs for a Target Locus Objective: To select high-efficiency, specific gRNAs for SpCas9-mediated targeting of a gene of interest (GOI). Materials: Computer with internet access; target gene sequence (FASTA format). Procedure:
- Input Sequence: Retrieve the genomic sequence of your GOI, including ~500 bp flanking your target region.
- PAM Identification: For SpCas9, scan the sequence for all instances of the "NGG" PAM motif.
- gRNA Extraction: For each PAM, extract the 20 nucleotides immediately 5' upstream. This is your potential spacer sequence.
- Multi-Tool Scoring: Submit the list of spacer sequences to at least two independent gRNA design platforms (e.g., Broad Institute's GPP, Benchling, or IDT's design tool). Compile the on-target efficiency scores (typically 0-100 scale).
- Off-Target Analysis: For the top 5 candidates by on-target score, run a genome-wide off-target search using Cas-OFFinder (settings: up to 3 mismatches, DNA bulge size 0). Exclude gRNAs with predicted off-targets in coding or regulatory regions of other genes.
- Final Selection: Select 2-3 gRNAs with the highest on-target scores and minimal/benign off-target predictions for empirical testing.
Protocol 2: Empirical Validation of gRNA Efficiency via T7 Endonuclease I (T7EI) Assay Objective: To experimentally measure the editing efficiency of selected gRNAs in your cell system. Materials: Transfected/transduced cells, Genomic DNA extraction kit, PCR reagents, T7 Endonuclease I enzyme (NEB), agarose gel electrophoresis system. Procedure:
- Genomic DNA Extraction: 72 hours post-transfection with your Cas9/gRNA construct, harvest cells and extract genomic DNA.
- PCR Amplification: Design primers flanking your target site (amplicon size 400-800 bp). Perform PCR using high-fidelity polymerase.
- DNA Heteroduplex Formation: Purify PCR product. Using a thermocycler, denature and reanneal the DNA: 95°C for 5 min, ramp down to 85°C at -2°C/s, then to 25°C at -0.1°C/s. This allows formation of heteroduplexes between wild-type and edited strands.
- T7EI Digestion: Prepare reaction: 200 ng reannealed PCR product, 1µl T7EI (NEB #M0302L), 2µl NEBuffer 2.1 in 20µl total. Incubate at 37°C for 30 minutes.
- Analysis: Run digested product on a 2% agarose gel. Cleavage products indicate presence of indels. Calculate efficiency: (1 - sqrt(1 - (b+c)/(a+b+c))) * 100, where a=uncut band intensity, b and c=cut band intensities.
Protocol 3: Evaluating Cas Variant Performance at a Non-Canonical PAM Site Objective: To compare the editing efficiency of SpCas9 versus SpCas9-NG at a target site with an NG PAM. Materials: Plasmids encoding SpCas9 and SpCas9-NG; gRNA expression scaffold; cells amenable to transfection; NGS library prep kit. Procedure:
- Construct Assembly: Clone the same target-specific gRNA sequence (designed for an NG PAM site) into vectors expressing SpCas9 and SpCas9-NG.
- Cell Transfection: Co-transfect cells in parallel with each Cas/gRNA plasmid complex. Include a no-guide control.
- Harvest and Sequence: Harvest genomic DNA 72 hours post-transfection. PCR amplify the target locus from all samples.
- Next-Generation Sequencing (NGS): Prepare amplicon sequencing libraries and perform high-depth sequencing (>50,000x coverage).
- Data Analysis: Use bioinformatics tools (CRISPResso2, BATCH-GE) to align sequences to the reference and quantify indel frequencies. Compare the % indels induced by SpCas9 vs. SpCas9-NG at the NG PAM target.
Visualizations
Title: gRNA Selection and Validation Workflow
Title: Cas Variant Selection Based on Editing Goal
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-Cas Editing Optimization
| Reagent / Solution | Function in Protocol | Example Supplier/Product |
|---|---|---|
| High-Fidelity DNA Polymerase | Accurate amplification of target loci for validation and NGS. | NEB Q5, Thermo Fisher Platinum SuperFi II. |
| T7 Endonuclease I | Detects indel mutations by cleaving DNA heteroduplexes. | New England Biolabs (M0302). |
| Next-Generation Sequencing Kit | Prepares amplicon libraries for deep sequencing of target sites. | Illumina TruSeq, IDT xGen Amplicon. |
| Cas9/gRNA Expression Vector | Delivers CRISPR components to mammalian cells. | Addgene: pSpCas9(BB)-2A-Puro (PX459). |
| Base Editor Plasmid | Enables precise point mutations without DSBs. | Addgene: pCMV_BE4max. |
| Prime Editor Plasmid | Enables templated edits without DSBs. | Addgene: pCMV-PE2. |
| Genomic DNA Extraction Kit | Purifies high-quality DNA from cultured cells. | Qiagen DNeasy, Promega Wizard. |
| CRISPR Design Software | In silico design and scoring of gRNAs. | Benchling, CRISPRscan, Broad GPP. |
Within the framework of a broader thesis on CRISPR-Cas mediated directed evolution, the fidelity of genetic modifications is paramount. Directed evolution accelerates the development of biomolecules with desired traits, but off-target editing by CRISPR-Cas systems can introduce confounding mutations, skewing selection outcomes and leading to erroneous conclusions. This document provides application notes and detailed protocols for identifying, quantifying, and mitigating off-target effects to ensure the integrity of directed evolution experiments.
Quantifying Off-Target Landscapes: Current Data
Off-target rates vary significantly based on the CRISPR nuclease, guide RNA design, and delivery method. The following table summarizes key quantitative findings from recent studies.
Table 1: Comparative Off-Target Profiles of Major CRISPR Systems
| CRISPR System | Primary Nuclease | Typical On-Target Efficiency | Reported Off-Target Rate (Range) | Key Determinants of Fidelity |
|---|---|---|---|---|
| CRISPR-Cas9 | SpCas9 | 70-90% | 0.1% - >50%* | Guide specificity, sgRNA secondary structure, chromatin accessibility, NGG PAM requirement. |
| High-Fidelity Cas9 | SpCas9-HF1, eSpCas9 | 50-80% | Often below detection limits (<0.1%) | Mutations reducing non-specific DNA contacts. |
| CRISPR-Cas12a | AsCas12a, LbCas12a | 60-85% | Generally lower than SpCas9 | Shorter guide (crRNA), T-rich PAM, staggered cut. |
| Base Editors | Cas9 nickase-deaminase fusions | Varies by base editor | 0.1% - 10% (dependent on window) | Deaminase activity window, guide-independent off-targets (RNA, ssDNA). |
| Prime Editors | Cas9 nickase-reverse transcriptase fusions | 10-50% (varies by edit) | Extremely low (<0.1%) reported | Dual guide requirement, reverse transcription template specificity. |
*Rate highly dependent on target and prediction method. Can be >50% for problematic guides.
Table 2: Off-Target Detection Method Sensitivities
| Method | Detection Principle | Sensitivity | Throughput | Key Limitation |
|---|---|---|---|---|
| Whole Genome Sequencing (WGS) | Sequencing of entire genome. | High (theoretical single-cell) | Low | Cost, data complexity, may miss low-frequency events. |
| CIRCLE-seq / GUIDE-seq | In vitro or in vivo capture of cleaved genomic DNA. | Very High (near single-molecule) | Medium-High | CIRCLE-seq is in vitro; GUIDE-seq requires oligonucleotide integration. |
| Digenome-seq | In vitro digestion of genomic DNA and WGS. | High | Medium | In vitro conditions may not reflect cellular chromatin state. |
| BLISS / SITE-seq | Direct labeling and sequencing of double-strand break sites. | High | High | Requires complex library preparation. |
Protocols for Ensuring Fidelity
Protocol 1:In SilicoGuide RNA Design and Selection for Directed Evolution Libraries
Objective: To computationally select sgRNAs with maximal predicted on-target activity and minimal off-target potential for targeting gene libraries in directed evolution.
Materials:
- Target gene sequence(s).
- Reference genome of host organism.
- gRNA design software (e.g., CHOPCHOP, Benchling, CRISPick).
Procedure:
- Input: Provide the FASTA sequence of the target gene(s) to be diversified.
- Parameter Setting:
- Set the appropriate PAM sequence (e.g., NGG for SpCas9).
- Select the "High-Fidelity" or "Specificity" scoring algorithm if available.
- Set guide length (typically 20nt).
- Run Analysis: The tool will output all possible sgRNAs with scores for on-target efficiency and off-target counts.
- Selection Criteria:
- Prioritize sgRNAs with high on-target scores (e.g., >60).
- Exclude any sgRNA with predicted off-target sites in:
- Protein-coding exons of other genes.
- Known regulatory elements (promoters, enhancers).
- Essential genes.
- For directed evolution, consider targeting conserved functional domains to maximize phenotypic impact.
- Final Validation: Cross-reference the top 3-5 candidate sgRNA sequences against the host genome using a basic alignment tool (BLAST) for a final specificity check.
Protocol 2: Experimental Validation of Off-Targets Using Targeted Deep Sequencing
Objective: To empirically measure off-target editing at predicted and discovered loci following CRISPR-mediated diversification.
Materials:
- Genomic DNA from edited and control cell pools.
- PCR primers for on-target and predicted off-target loci.
- High-fidelity PCR master mix.
- Next-generation sequencing library prep kit and access to a sequencer.
Procedure:
- Locus Amplification:
- Design PCR primers (~150-250 bp amplicon) flanking the on-target site and the top 10-20 in silico predicted off-target sites.
- Amplify each locus from treated and untreated control genomic DNA using a high-fidelity polymerase.
- Sequencing Library Preparation:
- Barcode the amplicons for multiplexed sequencing.
- Pool amplicons and prepare the library according to Illumina or equivalent protocols.
- Sequence to a depth of >100,000x reads per amplicon.
- Data Analysis:
- Demultiplex reads and align to the reference amplicon sequence.
- Use a variant-calling algorithm (e.g., CRISPResso2, BATCH-GE) to quantify insertion/deletion (indel) frequencies at the cut site.
- Calculate the percentage of reads containing indels for each locus.
- Interpretation:
- On-target efficiency = % indels at the primary target.
- Off-target activity = % indels at any other locus significantly above background (e.g., >0.1% and statistically significant vs. control).
Protocol 3: Implementing High-Fidelity Nucleases in Directed Evolution Workflows
Objective: To replace standard SpCas9 with a high-fidelity variant to reduce off-target background during library creation.
Materials:
- Plasmid expressing high-fidelity Cas9 (e.g., SpCas9-HF1, HypaCas9) or mRNA thereof.
- sgRNA expression construct (plasmid or synthetic).
- Delivery reagent (e.g., electroporation kit, lipid transfection reagent).
Procedure:
- Nuclease Cloning: Subclone the gene for the high-fidelity nuclease into your standard CRISPR delivery vector, replacing the wild-type Cas9. Alternatively, procure validated plasmids from addgene.
- Co-delivery: Co-transfect the high-fidelity nuclease expression construct and the sgRNA construct into your host cells alongside any donor library for homology-directed repair (HDR).
- Titration: Titrate the amount of nuclease plasmid/sgRNA to find the optimal balance between on-target HDR efficiency and cell viability. High-fidelity mutants often require precise stoichiometry.
- Validation: Follow Protocol 2 to confirm reduced off-target editing compared to the wild-type nuclease control under your specific experimental conditions.
Visualizations
Off-Target Mitigation in Directed Evolution Workflow
High-Fidelity vs. Wild-Type Cas9 Binding
The Scientist's Toolkit
Table 3: Essential Research Reagents for Off-Target Analysis
| Reagent / Material | Function / Purpose | Example Product / Note |
|---|---|---|
| High-Fidelity Cas9 Expression Plasmid | Expresses engineered nuclease variant with reduced non-specific DNA binding, lowering off-target effects. | Addgene #72247 (SpCas9-HF1), #114292 (HypaCas9). |
| Synthetic sgRNA (chemically modified) | Enhanced stability and reduced immune response; often paired with RNP delivery for reduced off-targets. | Truncated gRNAs (tru-gRNAs) or with 2'-O-methyl 3' phosphorothioate modifications. |
| Alt-R S.p. HiFi Cas9 Nuclease V3 | Purified protein for Ribonucleoprotein (RNP) complex formation. Direct delivery reduces vector persistence, improving specificity. | Integrated DNA Technologies (IDT). |
| GUIDE-seq Kit | All-in-one kit for unbiased in vivo off-target discovery via integration of a double-stranded oligodeoxynucleotide tag. | Available from various NGS service providers or as a published protocol. |
| CRISPResso2 Software | Computational pipeline for quantifying genome editing outcomes from deep sequencing data, including off-target analysis. | Open-source tool for batch analysis. |
| Next-Generation Sequencing Kit | For preparing targeted amplicon or whole-genome libraries to sequence edited genomic regions. | Illumina Nextera XT, Swift Biosciences Accel-NGS. |
| Control gRNA | Validated positive control (targeting a housekeeping gene) and negative control (non-targeting) gRNAs for experimental normalization. | Essential for benchmarking on/off-target ratios. |
Thesis Context: Within a CRISPR-Cas mediated directed evolution framework, isolating and characterizing rare gain-of-function variants from vast mutant libraries is a fundamental challenge. The efficacy of the entire evolutionary cycle depends on high-throughput, high-fidelity screening to accurately discriminate true signal from background noise.
Core Challenges & Quantitative Metrics
Effective screening for rare variants necessitates optimization of both throughput (number of variants assessed) and signal-to-noise ratio (SNR) (enrichment of true positives over background). The table below summarizes key performance metrics and targets for an ideal screening platform.
Table 1: Key Performance Targets for Rare Variant Screening
| Metric | Definition | Challenging Baseline | Improved Target |
|---|---|---|---|
| Library Diversity | Unique variants screened | 10^5 - 10^6 | >10^7 |
| Variant Frequency | Minimum detectable allele frequency | 0.1% - 1% | <0.01% |
| Assay Dynamic Range | Log difference between min/max signal | 10^2 - 10^3 | >10^4 |
| False Positive Rate (FPR) | Non-functional variants called positive | 1% - 5% | <0.1% |
| False Negative Rate (FNR) | Functional variants missed | 10% - 30% | <5% |
| Screening Throughput | Cells/variants processed per run | 10^7 - 10^8 cells | >10^9 cells |
Key Methodologies for Enhanced SNR & Throughput
Protocol 2.1: FACS-Based Enrichment with CRISPR Barcoding
Objective: Physically isolate rare variant-bearing cells based on a functional phenotype (e.g., surface expression, enzymatic activity) with high specificity.
Detailed Protocol:
- Library Construction & Delivery: Generate a pooled CRISPR activation (CRISPRa) or base-editor library targeting your gene set of interest. Include a unique, transcribed molecular barcode for each gRNA. Transduce at a low MOI (<0.3) into the target cell line to ensure single variant integration.
- Selection Pressure: Apply the relevant selective pressure (e.g., cytokine stimulation, low nutrient media, drug titration) for 5-14 days to allow phenotypic divergence.
- Staining & Sorting: a. Harvest cells and stain with fluorescent antibodies or activity-based probes targeting the phenotype of interest. b. Include a viability dye (e.g., DAPI) to exclude dead cells. c. Perform FACS using a high-speed sorter (e.g., Sony SH800, BD FACSAria III). Set stringent gates based on high-fluorescence controls (positive) and non-targeting gRNA controls (negative). d. Collect the top 0.1-1% of the fluorescent population as the "enriched" pool and a matched number of cells from the bulk/low-fluorescence population as a control.
- Barcode Amplification & Sequencing: Genomically extract DNA from both sorted pools. Amplify the integrated barcode region via PCR with Illumina-compatible primers. Sequence on a MiSeq or NextSeq platform.
- Analysis: Calculate gRNA enrichment in the high vs. low pool using tools like MAGeCK or BAGEL2. Significant outliers represent rare variant hits.
Protocol 2.2: Survival-Based Selection with Long-Term Culture
Objective: Enrich for variants conferring a proliferative advantage (e.g., drug resistance, improved fitness) over extended time.
Detailed Protocol:
- Competitive Pooled Culture: Transduce the CRISPR variant library into cells and maintain in culture at a minimum coverage of 500x (e.g., 5x10^7 cells for a 10^5 variant library).
- Longitudinal Sampling: At defined intervals (e.g., days 3, 7, 14, 21), harvest and cryopreserve ~1x10^7 cells per time point as a genomic DNA source.
- Deep Sequencing & Tracking: Extract gDNA from each time point. Amplify and sequence the variant barcodes to high depth (>1000x per variant). The relative frequency of each barcode over time reveals its fitness effect.
- SNR Enhancement: Spike-in a known ratio of control gRNAs (non-targeting, essential gene targeting) to normalize for sequencing noise and drift. Use the coefficient of variation (CV) of non-targeting guides to define the noise floor.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for High-Throughput Variant Screening
| Reagent/Material | Function & Critical Feature |
|---|---|
| CRISPRa/dCas9-VPR Lentiviral Library | Enables programmable transcriptional activation of endogenous genes to create gain-of-function variant pools. |
| Saturation Base Editor Library (e.g., A3A-BE3) | Creates all possible point mutations (C->T, G->A) within a target genomic window for dense variant scanning. |
| Unique Molecular Identifier (UMI) Barcodes | Integrated into library constructs to tag each variant, allowing for absolute quantification and reducing PCR/sequencing noise. |
| High-Sensitivity FACS Antibody/Probe | Crucial for phenotypic detection of rare cells; requires high specificity and brightness (e.g., PE/Cy7, Brilliant Violet 421). |
| Next-Generation Sequencing Kit (Illumina) | For deep, quantitative barcode sequencing. Low error rate is essential for accurate variant frequency calls. |
| Cell Recovery Medium | Used post-FACS to improve viability of sorted single cells, ensuring successful outgrowth for downstream validation. |
| Magnetic Bead-based gDNA Cleanup Kits | Enables rapid, high-throughput purification of genomic DNA from many cell samples for parallel barcode amplification. |
| Spike-in Control gRNA Plasmids | Known neutral and positive control gRNAs added at defined ratios to monitor selection efficiency and normalize screen data. |
Visualized Workflows & Logical Relationships
Title: Screening Strategy Selection Workflow
Title: Noise Sources and SNR Strategies
Title: End-to-End Pooled Screening Pipeline
In the context of CRISPR-Cas mediated directed evolution, the process of screening mutagenized libraries for enhanced phenotypes is fundamentally dependent on high-throughput sequencing and robust bioinformatic analysis. The transition from raw Next-Generation Sequencing (NGS) reads to a shortlist of high-confidence, beneficial mutations represents a critical, multi-step analytical pipeline. This protocol details the best practices for this data analysis workflow, ensuring accurate variant calling, functional annotation, and statistical validation within a directed evolution study.
Experimental Protocol: NGS Library Preparation & Sequencing for Variant Detection
Objective: To generate high-quality sequencing data from a pooled CRISPR-Cas edited cell population or organism library for variant identification and frequency calculation.
Materials:
- Genomic DNA or amplicon library from the pooled, selected population.
- Library preparation kit (e.g., Illumina DNA Prep).
- Target-specific or whole-genome sequencing primers.
- High-fidelity PCR mix.
- SPRI beads for size selection and cleanup.
- Qubit fluorometer and Bioanalyzer/TapeStation for QC.
- Compatible NGS platform (e.g., Illumina NovaSeq, MiSeq).
Methodology:
- Fragmentation & End Repair: Fragment gDNA to ~350 bp using enzymatic or acoustic shearing. Repair ends and add 'A' overhangs.
- Adapter Ligation: Ligate indexed, flow-cell compatible adapters to fragments. Perform dual indexing to allow multiplexing.
- Size Selection: Use SPRI bead-based cleanup to select fragments in the desired size range (e.g., 300-500 bp).
- Library Amplification: Perform limited-cycle PCR to enrich for adapter-ligated fragments.
- Quality Control: Quantify library concentration via Qubit and assess size distribution via Bioanalyzer. Pool libraries at equimolar ratios.
- Sequencing: Load pool onto sequencer. Aim for >100x average coverage across the target region to sensitively detect low-frequency variants.
Data Analysis Workflow: A Step-by-Step Protocol
Step 1: Raw Read Processing & Quality Control
- Tool: FastQC, MultiQC, Trimmomatic/FASTP.
- Protocol:
- Run FastQC on raw FASTQ files for per-base sequence quality, adapter content, GC distribution.
- Aggregate reports with MultiQC.
- Trim low-quality bases (Phred score <20), remove adapters, and discard short reads (<50 bp) using Trimmomatic or FASTP.
- Re-run FastQC on trimmed reads to confirm QC improvement.
Step 2: Alignment to Reference Genome
- Tool: BWA-MEM, Bowtie2, or HISAT2.
- Protocol:
- Index the reference genome/amplicon sequence using the chosen aligner (e.g.,
bwa index). - Align trimmed reads to the reference (
bwa mem). - Convert SAM output to sorted, indexed BAM files using SAMtools (
samtools sort,samtools index).
- Index the reference genome/amplicon sequence using the chosen aligner (e.g.,
Step 3: Post-Alignment Processing & Variant Calling
- Tool: GATK, SAMtools mpileup, bcftools.
- Protocol (GATK Best Practices):
- Mark Duplicates: Use Picard or GATK
MarkDuplicatesto flag PCR duplicates. - Base Quality Score Recalibration (BQSR): Generate recalibration table based on known variant sites and apply it to correct systematic sequencing errors.
- Variant Calling: For directed evolution, use a pooled calling approach.
- Use
HaplotypeCallerin-ERC GVCFmode on your pooled sample. - Alternatively, for a simpler pipeline, use
samtools mpileup -B -Q 20piped intobcftools call -mv -Ozto call variants.
- Use
- Filter raw variants based on quality metrics (e.g., QD > 2.0, FS < 60.0, SOR < 3.0, MQ > 40.0 for SNPs).
- Mark Duplicates: Use Picard or GATK
Step 4: Annotation & Functional Prediction
- Tool: SnpEff, SnpSift, VEP (Variant Effect Predictor).
- Protocol:
- Annotate the filtered VCF file with SnpEff using a custom-built database for your organism (
snpeff -v organism). - Annotations include: gene name, variant type (missense, nonsense, silent), amino acid change, and predicted impact (HIGH, MODERATE, LOW).
- (Optional) Use dbNSFP via SnpSift to add in silico prediction scores (e.g., SIFT, Polyphen2, CADD) for missense variants.
- Annotate the filtered VCF file with SnpEff using a custom-built database for your organism (
Step 5: Statistical Enrichment Analysis for Beneficial Mutants
- Tool: Custom R/Python scripts.
- Protocol:
- Calculate variant allele frequencies (VAF) in the selected population from the VCF.
- Compare to VAFs from the pre-selection or control population (essential control).
- Perform statistical testing (e.g., Fisher's Exact Test or a Binomial test) for each variant to assess significant enrichment.
- Apply multiple testing correction (e.g., Benjamini-Hochberg FDR < 0.05).
- Rank candidates by both statistical significance (FDR) and magnitude of enrichment (Fold-Change in VAF).
Data Presentation: Key Metrics & Results
Table 1: Post-Sequencing Quality Control Metrics
| Sample | Raw Reads | Q30 (%) | Adapter % | Trimmed Reads | Alignment Rate (%) | Mean Coverage |
|---|---|---|---|---|---|---|
| Pre-Selection Lib | 50,000,000 | 92.5 | 0.8 | 48,500,000 | 98.2 | 500x |
| Selected Pool (R1) | 45,000,000 | 93.1 | 0.5 | 44,200,000 | 98.7 | 450x |
| Selected Pool (R2) | 47,000,000 | 92.8 | 0.6 | 46,100,000 | 98.5 | 470x |
Table 2: Top Enriched Variants from Directed Evolution Screen
| Gene | Nucleotide Change | Amino Acid Change | VAF Pre-Select | VAF Post-Select | Fold-Change | FDR p-value | Predicted Impact |
|---|---|---|---|---|---|---|---|
| TARGET_A | c.742C>T | p.Arg248Trp | 0.0005 | 0.125 | 250.0 | 1.2e-15 | MODERATE (Missense) |
| TARGET_A | c.1102G>A | p.Gly368Ser | 0.0007 | 0.098 | 140.0 | 4.5e-12 | MODERATE (Missense) |
| REGULATOR_B | c.88_89delAT | p.Met30Valfs*12 | 0.0003 | 0.045 | 150.0 | 2.1e-09 | HIGH (Frameshift) |
Visualization of Workflows
Title: Data Analysis Pipeline from NGS to Mutations
Title: Protocol Context in Directed Evolution Thesis
The Scientist's Toolkit: Research Reagent & Software Solutions
Table 3: Essential Tools for NGS Data Analysis in Directed Evolution
| Category | Item/Software | Function/Brief Explanation |
|---|---|---|
| Sequencing Service | Illumina DNA Prep Kit | Library preparation for whole-genome or targeted sequencing. |
| Alignment | BWA-MEM (v0.7.17+) | Efficient alignment of short sequencing reads to a reference genome. |
| File Processing | SAMtools/BEDTools | Manipulation, sorting, indexing, and intersection of alignment files. |
| Variant Calling | GATK (v4.0+) | Industry-standard toolkit for variant discovery with robust filtering. |
| Variant Annotation | SnpEff (v5.0+) | Rapid annotation of genetic variants and prediction of functional effects. |
| Statistical Analysis | R/Bioconductor (DESeq2 edgeR) | Statistical testing for variant enrichment between populations. |
| Data Visualization | Integrative Genomics Viewer (IGV) | Visual exploration of aligned reads and called variants in genomic context. |
| Workflow Management | Nextflow/Snakemake | Orchestration of complex, reproducible bioinformatic pipelines. |
Benchmarking Success: Validating Results and Comparing Methodologies
Within a CRISPR-Cas mediated directed evolution workflow, the generation of variant libraries is merely the first step. The critical, and often bottleneck, phase is the functional validation of evolved protein sequences. A robust validation framework is essential to distinguish genuine improvements from neutral or destabilizing mutations, ensuring that selected variants meet the desired functional and biophysical criteria for downstream applications in therapeutics and industrial biocatalysis.
Core Validation Pillars
A comprehensive framework rests on four pillars: Expression & Solubility, Thermodynamic Stability, Functional Activity, and Conformational Integrity. Assays within each pillar provide complementary data, building a holistic profile of the evolved protein.
Table 1: Pillars of the Protein Validation Framework
| Validation Pillar | Primary Objective | Key Quantitative Metrics | Common Assay Techniques |
|---|---|---|---|
| Expression & Solubility | Assess production yield and fraction of properly folded protein. | - Total protein yield (mg/L)- Soluble fraction (%)- Aggregation propensity | SDS-PAGE, Western Blot, Solubility assays (e.g., centrifugation + Bradford) |
| Thermodynamic Stability | Measure resistance to thermal/chemical denaturation. | - Melting Temperature (Tm, °C)- ΔG of unfolding (kJ/mol)- [C]₁/₂ of denaturant (M) | Differential Scanning Fluorimetry (DSF), Differential Scanning Calorimetry (DSC), Chemical Denaturation (CD/Fluorescence) |
| Functional Activity | Quantify catalytic efficiency or binding affinity. | - kcat/KM (M⁻¹s⁻¹)- IC₅₀ (nM)- K_D (nM) | Enzyme kinetics (SPR, HPLC), MIC assays (antibiotics), Binding assays (ELISA, SPR) |
| Conformational Integrity | Verify correct higher-order structure and dynamics. | - Secondary structure content (%)- RMSD (Å) from model- Thermal aggregation onset (°C) | Circular Dichroism (CD), Size Exclusion Chromatography (SEC), Analytical Ultracentrifugation (AUC) |
Detailed Application Notes & Protocols
Protocol: High-Throughput Thermal Stability via DSF
Context: Following a CRISPR-Cas mediated evolution campaign for enhanced protease stability, screen 100s of variants for improved Tm.
- Reagents: Purified protein variants (0.2-0.5 mg/mL in PBS, pH 7.4), SYPRO Orange dye (5000X stock in DMSO), sealing foil for microplates.
- Equipment: Real-time PCR instrument with HRM capability, 96- or 384-well PCR plates.
- Procedure:
- Prepare a 10X working stock of SYPRO Orange in PBS.
- In each well, mix 18 µL of protein sample with 2 µL of 10X SYPRO Orange dye. Include a buffer-only control.
- Seal plate, centrifuge briefly.
- Run in RT-PCR instrument: Ramp from 25°C to 95°C at 1°C/min, with fluorescence acquisition (ROX/FAM filter set) at each step.
- Data Analysis: Plot fluorescence (F) vs. Temperature (T). Calculate Tm as the inflection point using the first derivative (dF/dT). A >2°C increase over wild-type is typically significant.
Protocol: Determining Catalytic Efficiency (kcat/KM) for an Evolved Enzyme
Context: Validate the activity of an evolved transglycosylase from a CRISPR-Cas targeted library.
- Reagents: Purified enzyme variants, chromogenic/fluorogenic substrate (e.g., pNPG for glycosidases), assay buffer (optimal pH).
- Equipment: Microplate reader, 96-well flat-bottom plates, precision pipettes.
- Procedure (Initial Rate Method):
- Prepare a substrate dilution series (typically 8 concentrations spanning 0.2-5 x K_M).
- Dilute enzyme to a concentration that yields linear progress curves for at least 2 minutes.
- In a plate, add buffer and substrate solution. Start reaction by adding enzyme (final volume 100 µL).
- Immediately initiate kinetic read (e.g., absorbance at 405 nm for pNP) every 10 seconds for 3 minutes.
- Repeat in triplicate.
- Data Analysis: Calculate initial velocity (V₀) for each [S]. Fit data to the Michaelis-Menten equation (V₀ = (Vmax * [S]) / (KM + [S])) using non-linear regression (e.g., GraphPad Prism). Calculate kcat = Vmax / [Enzyme]. Report kcat/KM.
Protocol: Conformational Analysis via Circular Dichroism (CD)
Context: Confirm that an evolved antibody fragment (scFv) with improved affinity retains its native β-sheet fold.
- Reagents: Purified protein in low-absorbance buffer (e.g., 5 mM phosphate, pH 7.0), 0.1 µm filtered.
- Equipment: Jasco or Applied Photophysics Chirascan spectropolarometer, quartz cuvette (pathlength 0.1 cm for far-UV).
- Procedure (Far-UV Scan):
- Dialyze protein into CD buffer extensively. Determine accurate concentration (A₂₈₀).
- Load sample (typical requirement: 0.1-0.2 mg/mL in 200 µL) into cuvette.
- Acquire spectrum from 260 nm to 190 nm at 20°C, with 1 nm bandwidth, 1 s response time.
- Subtract buffer baseline spectrum.
- Data Analysis: Convert raw ellipticity (mdeg) to mean residue ellipticity [θ]. Compare spectral minima/maxima positions (e.g., ~218 nm & ~195 nm for β-sheet) to wild-type. Use deconvolution algorithms (SELCON3, CONTIN) to estimate secondary structure percentages.
Visualizing the Validation Workflow
Diagram Title: Protein Validation Workflow Post-Directed Evolution
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Validation Assays
| Reagent/Material | Supplier Examples | Function in Validation |
|---|---|---|
| SYPRO Orange Dye | Thermo Fisher, Sigma-Aldrich | Environment-sensitive fluorescent dye for DSF; binds hydrophobic patches exposed during protein unfolding. |
| Precision Protease Kits | Roche, Qiagen, NEB | For limited proteolysis assays to probe conformational rigidity and flexible regions. |
| Chromogenic/Fluorogenic Substrates | Sigma-Aldrich, Cayman Chemical, Bachem | Enable direct, continuous measurement of enzyme activity (e.g., pNP-linked sugars, AMC fluorophores). |
| HisTrap HP Columns | Cytiva | Standardized immobilized metal affinity chromatography (IMAC) for high-yield purification of His-tagged evolved variants. |
| Stability & Storage Buffers | Hampton Research, Molecular Dimensions | Pre-formulated, optimized buffers for crystallization and long-term stability studies. |
| SEC Standards | Agilent, Bio-Rad | Molecular weight marker kits for calibrating Size Exclusion Chromatography to assess monomericity/aggregation. |
| SPR Sensor Chips (CM5) | Cytiva | Gold-standard for label-free, real-time kinetic analysis of binding interactions (KD, kon, k_off). |
| CD Calibration Standard (Ammonium d-10-Camphorsulfonate) | JASCO, Avantor | Essential for verifying the wavelength accuracy and amplitude calibration of a CD spectropolarimeter. |
Application Note 1: Directed Evolution of Thermostable T7 RNA Polymerase Using CRISPR-Cas9
Background
This application highlights a CRISPR-Cas9-facilitated continuous evolution platform (EvolvR) applied to enhance the thermostability of T7 RNA polymerase, a critical enzyme for in vitro transcription. The goal was to generate variants functional at elevated temperatures for robust, high-yield mRNA synthesis, a key process in therapeutic mRNA production.
Key Quantitative Results
Table 1: Thermostability and Activity of Evolved T7 RNA Polymerase Variants
| Variant ID | Melting Temp (Tm) Δ (°C) | Half-life at 50°C (min) | Relative Activity at 45°C (%) | Key Mutations |
|---|---|---|---|---|
| Wild-Type | 0 (ref: 47.5°C) | 12 ± 2 | 100 (ref) | N/A |
| EV-T7-1 | +3.2 | 35 ± 5 | 98 ± 5 | S430P, N433T |
| EV-T7-5 | +5.7 | 75 ± 8 | 120 ± 10 | F849I, S430P |
| EV-T7-12 | +8.1 | 210 ± 15 | 105 ± 7 | S430P, N433T, F849I, H300Q |
Experimental Protocol
Protocol 1.1: CRISPR-Cas9-Mediated Continuous Diversification and Selection for Thermostability
Objective: To generate and select T7 RNA polymerase variants with increased thermostability using the EvolvR system.
Materials:
- E. coli strain harboring the EvolvR system: nCas9 (H840A)-dCas9-PolI3M fusion, gRNA plasmid targeting the T7 pol gene locus.
- Induction Media: LB + 0.2% L-Arabinose (to induce EvolvR nicking/diversification) + Anhydrotetracycline (aTc, for gRNA expression).
- Thermal Challenge Plates: Agar plates with inducer for T7 polymerase-dependent reporter (e.g., GFP under T7 promoter), incubated at elevated temperature (45-50°C).
- Sequence Capture: Q5 High-Fidelity DNA Polymerase, T7 specific primers, NGS library prep kit.
Procedure:
- Library Generation: Grow the E. coli EvolvR-T7 strain in induction media for 12-16 hours (approx. 60 generations) to allow continuous random mutagenesis within the ~200 bp window targeted by the gRNA.
- Thermal Pre-Selection: Dilute the culture and plate on thermal challenge plates. Incubate at permissive temperature (37°C) for 4 hours, then shift to restrictive temperature (e.g., 48°C) for 24-48 hours. Only cells expressing sufficiently stable and active T7 polymerase will activate the reporter and form colonies.
- Colony Screening: Pick surviving colonies, inoculate into deep-well plates with liquid challenge media, and perform a kinetic fluorescence assay at high temperature to quantify activity.
- Variant Recovery: Isolate plasmid DNA from top performers. Amplify the T7 pol gene via PCR and sequence.
- Validation: Clone identified variant genes into a clean expression vector, transform into naive E. coli. Purify the protein and characterize biophysically (DSC for Tm, activity assays at temperature gradients).
Application Note 2: EnhancingBacillus subtilisLipase A Activity and Stability via Base Editor-Driven Saturation Mutagenesis
Background
This case study utilizes a CRISPR-Cas9-derived cytidine base editor (CBE) for targeted, multiplexed saturation mutagenesis to improve the catalytic efficiency and solvent stability of Bacillus subtilis Lipase A (BSLA), an industrial biocatalyst.
Key Quantitative Results
Table 2: Biochemical Properties of Base-Edited BSLA Variants
| Variant ID | Specific Activity (U/mg) Δ% | kcat/Km (s⁻¹M⁻¹x10⁴) | Solvent Stability (t½ in 25% DMSO, min) | Thermostability (T50, °C) | Key Mutations (C→T edits) |
|---|---|---|---|---|---|
| WT BSLA | 0% (ref: 450 U/mg) | 1.5 ± 0.2 | 30 ± 5 | 45.2 | N/A |
| BE-BSLA-3 | +85% | 2.9 ± 0.3 | 55 ± 7 | 47.5 | Q12L, A20T |
| BE-BSLA-7 | +210% | 4.8 ± 0.5 | 120 ± 15 | 51.8 | A20T, P94S, D133G |
| BE-BSLA-11 | +175% | 4.1 ± 0.4 | 240 ± 20 | 53.1 | P94S, D133G, N166Y |
Experimental Protocol
Protocol 2.1: Multiplexed Base Editor Saturation Screening for Lipase Engineering
Objective: To simultaneously introduce targeted C-to-T (resulting in specific amino acid) mutations at multiple predefined codons in the bsla gene and screen for improved activity and stability.
Materials:
- B. subtilis strain with integrated CBE (nCas9-DdCBE-UGI) and a repair template for library construction.
- gRNA Pool: A plasmid library expressing gRNAs targeting 5-8 specific codons in the bsla gene for conversion.
- Screening Plates: Tributyrin agar plates for halo assay (activity), plates with sub-lethal DMSO concentration (stability).
- Microplate Assay Buffer: p-Nitrophenyl butyrate (pNPB) in isopropanol, 50 mM Tris-HCl pH 8.0.
Procedure:
- Library Transformation: Co-transform the pool of gRNA plasmids into the B. subtilis CBE strain. Grow under selection to allow base editing at all target sites across the population.
- Primary High-Throughput Screening: Plate the transformation on tributyrin agar plates. Incubate at 37°C for 48 hours. Colonies with higher lipase activity will display larger hydrolysis halos.
- Secondary Stability Screening: Inoculate colonies from the primary screen into 96-well deep plates containing media with 15% DMSO. Grow for 24h, then spot culture supernatant on fresh tributyrin plates. Variants with improved solvent stability show larger halos post-challenge.
- Quantitative Characterization: Express and purify hits from a heterologous host (E. coli). Measure specific activity using pNPB hydrolysis assay (A410). Determine T50 (temperature at which 50% activity is lost after 10 min incubation) and DMSO half-life.
- Data Analysis: Correlate NGS data of pre- and post-selection populations with phenotypic data to identify beneficial mutation combinations.
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for CRISPR-Cas Directed Evolution of Proteins
| Reagent / Material | Function in Experiment | Example Product/Catalog |
|---|---|---|
| nCas9 (H840A/D10A) Nickase | Creates single-strand breaks in target DNA, enabling high-efficiency homology-directed repair (HDR) or triggering continuous mutagenesis (EvolvR). | Addgene #41816 (pEvolvR) |
| Cytidine Base Editor (CBE) | Converts C•G to T•A base pairs within a defined window without requiring double-strand breaks or donor templates, enabling precise saturation mutagenesis. | Addgene #100812 (pCMV-BE4) |
| Error-Prone DNA Polymerase Variant | Used in EvolvR systems as the mutagenic fusion to nCas9. Introduces random mutations during gap repair at the nicked site. | E. coli PolI3M (mutant) |
| gRNA Expression Library Plasmid Pool | Delivers a pool of target-specific guide RNAs to direct Cas9 variants to multiple genomic loci or codons simultaneously for multiplexed evolution. | Custom synthesized array oligo pool cloned into gRNA scaffold vector. |
| Reporter Plasmid with Conditional Survival/Output | Links desired protein property (e.g., thermostability, activity) to cell survival or fluorescence, enabling powerful positive selection. | Plasmid with antibiotic resistance gene under control of target protein-dependent promoter. |
| Temperature-Controlled Incubator/Shaker | Essential for applying thermal stress during selection phases to enrich for thermostable protein variants. | Standard microbiological incubator with gradient temperature function. |
| Microplate Reader with Temperature Control | For high-throughput kinetic analysis of enzyme activity and stability under varying thermal or solvent conditions. | Tecan Spark, BioTek Synergy H1. |
| Next-Generation Sequencing (NGS) Kit | For deep sequencing of evolved gene libraries pre- and post-selection to identify enriched mutations and evolutionary pathways. | Illumina MiSeq, NovaSeq library prep kits. |
Visualization: Experimental Workflows and Pathways
Diagram 1: EvolvR workflow for thermostability.
Diagram 2: CBE mechanism for targeted mutagenesis.
Diagram 3: Thesis context integrating case studies.
Application Notes
Within the thesis exploring CRISPR-Cas-mediated directed evolution, this analysis provides a comparative framework for selecting mutagenesis and screening strategies. The core distinction lies in precision and throughput: CRISPR-based systems offer targeted, in vivo diversification of specific genomic loci, while classical methods like error-prone PCR (epPCR) and DNA shuffling provide broad, in vitro library generation for in vitro or plasmid-based evolution.
Table 1: Quantitative Comparison of Key Evolution Methods
| Parameter | CRISPR-Driven Evolution (e.g., CRISPR-X, DOMESTIC) | Error-Prone PCR (epPCR) | DNA Shuffling |
|---|---|---|---|
| Mutagenesis Mechanism | Targeted, Cas9-fused deaminase or reverse transcriptase | Random, polymerase misincorporation | Recombination of homologous sequences |
| Mutation Rate (Typical Range) | 10^-5 to 10^-3 per base (tunable, localized) | 0.1-2 mutations per kb per round | N/A (recombines existing variants) |
| Library Size (Practical) | Limited by host transformation (~10^9 in yeast/bacteria) | Very high (>10^12 in vitro) | High (10^10 - 10^12 in vitro) |
| Primary Library Location | In vivo (chromosomal) | In vitro (plasmid) | In vitro (gene fragments) |
| Key Advantage | In vivo, functional screening; targeted diversity | Simplicity, high randomness | Recombines beneficial mutations |
| Main Limitation | Lower library diversity, host-dependent | Primarily in vitro, non-targeted | Requires sequence homology |
| Best Suited For | Improving function of genomic pathways, membrane proteins, complex traits | Initial exploration of single-gene sequence space | Accelerating evolution of genes with known beneficial variants |
Experimental Protocols
Protocol 1: CRISPR-Driven Targeted Evolution using a Base Editor Fusion Objective: To evolve a specific gene in its native genomic context in S. cerevisiae for enhanced thermostability.
- Construct Design: Clone a cytidine deaminase (e.g., APOBEC1)-dCas9 fusion protein expression cassette into a yeast plasmid. Design sgRNA(s) targeting a ~100bp window within the gene of interest (GOI).
- Library Generation: Co-transform the fusion plasmid and sgRNA plasmid(s) into the yeast strain harboring the native GOI. Plate on selective media. The deaminase will introduce C>T (or G>A) mutations within the sgRNA-targeted window during cultivation.
- Screening: Apply selective pressure (e.g., elevated temperature). Surviving colonies are isolated. The genomic region of the GOI is PCR-amplified and sequenced to identify mutations.
- Iteration: Beneficial mutations can be fixed, and new sgRNAs designed to target adjacent regions for additional rounds.
Protocol 2: Error-Prone PCR and Plasmid Library Construction Objective: To create a random mutant library of a bacterial antibiotic resistance gene.
- epPCR Setup: In a 50µL reaction, combine: 10-100 ng template DNA, 1X proprietary mutagenesis buffer (e.g., with Mn2+), 0.2 mM each dNTP, 0.2 µM forward/reverse primers, 5 U Taq polymerase. Standard PCR cycling conditions are used.
- Purification: Clean the PCR product using a spin column kit.
- Cloning: Digest the purified PCR product and the recipient vector with appropriate restriction enzymes. Ligate and transform into ultra-competent E. coli.
- Library Harvest: Plate a fraction to estimate diversity. Harvest the remainder via plasmid prep from the pooled colonies.
Protocol 3: DNA Shuffling for Gene Family Recombination Objective: To recombine homologous sequences from multiple bacterial laccase genes.
- Fragment Preparation: Amplify target genes from diverse parents via standard PCR. Purify products.
- Fragmentation: Use DNase I in the presence of Mn2+ to randomly digest genes into 50-100 bp fragments.
- Reassembly PCR: Perform a primerless PCR: dilute fragments, add dNTPs and polymerase. Use cycling: 94°C (denaturation), 30-40 cycles of [94°C (30s), 50-55°C (30s), 72°C (30s)], 72°C (5min). Fragments prime each other based on homology.
- Amplification: Add outer primers and perform standard PCR to amplify full-length, reassembled genes.
- Cloning & Screening: Clone products into an expression vector, transform, and screen for activity.
Visualizations
Title: DNA Shuffling Workflow for Gene Recombination
Title: In Vivo CRISPR-Driven Directed Evolution Workflow
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Context |
|---|---|
| dCas9-Fusion Plasmid (e.g., pCRISPR-X) | Expresses catalytically dead Cas9 fused to a mutagenic enzyme (deaminase, reverse transcriptase). Enables targeted, in vivo mutagenesis. |
| Mutagenic PCR Kit (e.g., Genemorph II) | Optimized buffer systems with MnCl2 and unbalanced dNTPs to standardize and control error rates during epPCR. |
| Ultra-Competent Cells (e.g., NEB 10-beta) | High-efficiency transformation cells crucial for achieving large library sizes (>10^9 CFU/µg) from in vitro cloning steps. |
| Homologous Reassembly Enzyme Mix | Specialized polymerases/blends optimized for efficient reassembly of fragmented DNA during DNA shuffling protocols. |
| sgRNA Library Pool | A synthesized pool of guide RNAs targeting multiple regions of a gene, used to spread mutagenesis across a larger sequence space in CRISPR evolution. |
| Next-Gen Sequencing Kit (e.g., MiSeq) | For deep sequencing of mutant libraries pre- and post-selection to quantify enrichment and identify consensus mutations. |
Introduction Within the context of CRISPR-Cas mediated directed evolution (CDE), the comparative advantages and limitations over traditional methods (e.g., error-prone PCR, mutagenic strains, site-saturation libraries) define its transformative potential. This document details application notes and protocols for implementing CDE, focusing on its core operational parameters.
1. Quantitative Comparison: CDE vs. Traditional Methods
Table 1: Performance Metrics Comparison
| Parameter | CRISPR-Cas Directed Evolution (CDE) | Traditional Methods (e.g., Error-Prone PCR) |
|---|---|---|
| Speed | Weeks. Enables rapid, continuous, and recursive mutagenesis in situ without subcloning. | Months. Iterative cycles require sequence-verified subcloning, transformation, and screening for each round. |
| Scale | >10^9 variants per library. Can exploit large-scale pooled delivery via lentiviral transduction in mammalian systems. | ~10^6 - 10^8 variants. Limited by transformation efficiency (especially in mammalian cells) and plasmid library size. |
| Control | High. Mutagenesis is targeted to specific genomic loci or plasmid positions via gRNA design. Tunable mutation rates via modulation of repair template concentration. | Low. Mutagenesis is random across the entire gene of interest, requiring extensive screening to find beneficial mutations. |
| Targetability | Precise. Can evolve regulatory elements (promoters, enhancers), non-coding RNA, or specific protein domains with single-nucleotide precision. | Diffuse. Primarily suited for evolving coding sequences of plasmid-borne genes; targeting specific genomic regions or regulatory elements is highly inefficient. |
| Key Limitation | Efficiency dependent on HDR/cellular repair pathways; potential for indel formation; delivery complexity for primary cells. | Low frequency of beneficial mutations; high background of neutral/deleterious variants; cannot easily target genomic loci in native chromosomal context. |
2. Core Protocol: Continuous Evolution in Mammalian Cells using a CRISPR-Cas9 Base Editor System
This protocol enables rapid protein evolution through targeted, diversifying base editing at a defined locus.
- Objective: To generate and select for variants of a surface receptor (e.g., PD-1) with enhanced binding affinity in a human cell line.
- Workflow Diagram Title: CDE with Base Editor Workflow
Detailed Protocol Steps:
Day 1-3: Library Construction and Lentivirus Production
- Design: Synthesize an oligonucleotide pool encoding a library of gRNAs targeting the protein domain of interest. Clone into a lentiviral gRNA expression backbone (e.g., lentiGuide-Puro).
- Co-transfection: In HEK293T cells, co-transfect the gRNA library plasmid, the BE4max base editor plasmid (expressing a cytidine deaminase fused to nickase Cas9), and third-generation lentiviral packaging plasmids (psPAX2, pMD2.G) using a PEI-pro transfection reagent.
- Harvest Virus: Collect lentiviral supernatant at 48h and 72h post-transfection, filter through a 0.45 µm filter, and concentrate using Lenti-X Concentrator.
Day 4-5: Target Cell Transduction and Diversification
- Transduce: Transduce your target cell line (e.g., HEK293T expressing the target gene) with the concentrated lentivirus at an MOI of ~0.3 to ensure single gRNA integration. Include puromycin selection (2 µg/mL) starting 48h post-transduction for 3 days.
- Diversify: Cultivate the polyclonal pool for 7-10 days to allow base editor expression and generation of a diverse mutational library at the target locus. Base editing efficiency can be monitored by targeted NGS of an unselected sample.
Day 14-21: Selection and Analysis
- Apply Selection: Subject the diversified cell pool to your functional screen (e.g., incubate with fluorescently labeled ligand, then sort the top 1% highest-binding cells via FACS).
- Recover & Expand: Culture the sorted population for 7 days to expand.
- Harvest & Sequence: Extract genomic DNA from the pre-selection and post-selection populations using a DNeasy Blood & Tissue Kit. Perform PCR amplification of the target region and submit for next-generation sequencing.
- Analysis: Analyze sequencing data to identify mutations significantly enriched in the post-selection pool. Statistical analysis (e.g., using
MAGeCKorDESeq2for count data) is required.
3. The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for CRISPR-CDEX
| Reagent / Material | Function & Brief Explanation |
|---|---|
| Base Editor Plasmid (e.g., BE4max) | Engineered fusion protein (Cas9 nickase + cytidine deaminase + uracil glycosylase inhibitor). Enables direct, irreversible conversion of C•G to T•A base pairs without requiring double-strand breaks or donor templates. |
| Lentiviral gRNA Library Backbone | Allows for efficient, stable integration of the gRNA expression cassette into the host genome, enabling long-term expression and propagation of the library. |
| Lenti-X Concentrator | Polyethylene glycol-based solution for rapid, simple concentration of lentiviral particles, increasing viral titer for more efficient transduction. |
| PEI-pro Transfection Reagent | High-efficiency polymer for transient transfection of plasmid DNA into packaging cells for high-titer lentivirus production. |
| Next-Generation Sequencing Kit | For preparation of sequencing libraries from amplified genomic DNA to enable deep sequencing of the target locus pre- and post-selection. |
| FACS Aria or Similar Cell Sorter | Essential instrument for high-throughput, quantitative isolation of cell populations based on phenotypic readouts (e.g., binding affinity, fluorescence). |
4. Pathway Diagram: Logical Relationship in a CDE Cycle
Diagram Title: CDE Iterative Cycle Logic
This application note details protocols for integrating phage display, yeast display, and machine learning predictions within a CRISPR-Cas-mediated directed evolution pipeline. Directed evolution accelerates protein engineering, and CRISPR-Cas systems can precisely introduce diversity. These methods generate high-dimensional data, which, when coupled with ML models, enables predictive in silico evolution and intelligent library design for therapeutic and diagnostic applications.
Key Integrated Workflows
CRISPR-Cas Mediated Library Generation for Display Selection
CRISPR-Cas9 facilitates precise, multiplexed gene integration of variant libraries into the host genome for display technologies.
Protocol: CRISPR-Cas9 Mediated Library Integration for Yeast Surface Display
Objective: Integrate a pooled, mutagenized gene library into the Saccharomyces cerevisiae genome at the Aga2p display locus.
Materials:
- S. cerevisiae EBY100 strain.
- Plasmid library encoding mutant genes with homology arms (~500 bp) to the Aga2p locus.
- pCas9-gRNA plasmid targeting the safe-harbor Aga2p integration site.
- LiAc/SS carrier DNA/PEG transformation mix.
- Synthetic Complete (SC) media lacking appropriate amino acids for selection.
- Induction media: SG-CAA (galactose-containing) for expression.
Method:
- Design & Preparation: Design gRNA with minimal off-targets in yeast. Prepare the linear donor DNA library (mutant gene + homology arms) via pooled PCR.
- Co-transformation: Grow EBY100 to mid-log phase. Co-transform 1 µg of linear donor DNA library and 100 ng of pCas9-gRNA plasmid using a high-efficiency LiAc protocol.
- Selection & Induction: Plate transformations on SC-Trp/-Ura plates to select for both donor integration (e.g., Trp+) and Cas9 plasmid retention (Ura+). Incubate at 30°C for 48-72 hours.
- Library Induction: Pool colonies, inoculate into SR-CAA (glucose) media, grow overnight. Centrifuge and resuspend in SG-CAA media to induce Aga2p fusion protein expression. Incubate at 20°C, 250 rpm for 20-24 hours.
- Validation: Check library diversity via NGS of the integrated region from pooled genomic DNA.
Phage Display Biopanning with Enriched Libraries
Post-CRISPR evolution rounds, enriched pools can be cloned into phage vectors for finer selection.
Protocol: Phage Biopanning with Pre-Enriched Yeast Display Outputs
Objective: Isolate high-affinity binders from a phage library constructed from sequences enriched after prior yeast display selection rounds.
Materials:
- M13KE phage display vector.
- E. coli ER2738 host strain.
- Target antigen, immobilized on streptavidin-coated magnetic beads or Nunc MaxiSorp plates.
- PEG/NaCl for phage precipitation.
- Triethylamine (elution buffer).
Method:
- Library Construction: Amplify enriched gene pools from yeast genomic DNA. Clone into M13KE via restriction digestion/ligation or Gibson Assembly. Electroporate into ER2738 to create the primary phage library (>10^9 diversity).
- Biopanning: Incubate phage library (10^11 - 10^12 pfu) with immobilized antigen for 1h at RT. Wash extensively with PBST (0.1% Tween-20) to remove non-specific binders. Elute bound phage with 100 mM triethylamine (neutralize immediately) or via competitive elution with soluble target.
- Amplification & Iteration: Infect log-phase ER2738 with eluted phage, culture overnight, and precipitate amplified phage output with PEG/NaCl. Use as input for subsequent rounds (typically 3-4 rounds).
- Analysis: Sequence output from rounds 3 and 4. Titrate input, output, and amplified phage to calculate enrichment.
Machine Learning Model Training for Binding Prediction
Data from display campaigns train models to predict fitness, guiding future library design.
Protocol: Training a Neural Network on Display Sequencing Data
Objective: Train a model to predict binding fitness (e.g., enrichment score) from protein sequence.
Materials:
- High-throughput sequencing data from display selection input and output pools.
- Computational environment (Python, TensorFlow/PyTorch).
- Enrichment scores calculated per variant: Log2(Output frequency / Input frequency).
Method:
- Data Curation: Align sequencing reads to reference. Count variant frequencies in pre- (input) and post-selection (output) pools. Filter variants with low read counts (<10 in input).
- Feature Encoding: Encode amino acid sequences using one-hot encoding, physicochemical property embeddings, or learned embeddings.
- Model Architecture: Implement a convolutional neural network (CNN) for local pattern detection or a transformer model for long-range dependencies.
- Input: Encoded sequence matrix.
- Layers: 2-3 convolutional layers with ReLU, pooling, fully connected layers.
- Output: Single neuron for regression (predicted enrichment score).
- Training: Split data 80/10/10 (train/validation/test). Use Mean Squared Error loss and Adam optimizer. Apply early stopping based on validation loss.
- Validation: Evaluate on held-out test set. Correlate predicted vs. calculated enrichment scores. Use model to rank in silico designed mutants for experimental testing.
Table 1: Comparison of Display Technologies Integrated with CRISPR-Cas Evolution
| Feature | Yeast Surface Display | Phage Display |
|---|---|---|
| Display System | Eukaryotic, Aga1p-Aga2p agglutinin fusion | Prokaryotic, pIII or pVIII coat protein fusion |
| Library Size | 10^7 - 10^9 variants | 10^9 - 10^11 variants |
| CRISPR Integration | Direct genomic integration possible via homology-directed repair (HDR). | Typically plasmid-based; CRISPR used for in vivo mutagenesis in E. coli. |
| Selection Modality | FACS (quantitative, multiparametric) | Biopanning (affinity-based) |
| Expression Host | Saccharomyces cerevisiae (eukaryotic PTMs) | Escherichia coli (no eukaryotic PTMs) |
| Typical Throughput (Screening) | High (FACS: >10^8 cells/hour) | Medium (Sequencing output: 10^3 - 10^5 clones) |
| Key Metric | Mean Fluorescence Intensity (MFI) by FACS | Phage Titer (pfu) & Enrichment Ratio |
Table 2: ML Model Performance on Directed Evolution Data (Representative Benchmarks)
| Model Type | Training Data Source | Test Set R² (vs. Experimental Fitness) | Key Advantage |
|---|---|---|---|
| Convolutional Neural Network (CNN) | Yeast display FACS enrichment scores | 0.72 - 0.85 | Captures local motif importance. |
| Transformer (Protein Language Model) | Phage display NGS counts + UniRef corpus | 0.78 - 0.90 | Leverages evolutionary context; requires less project-specific data. |
| Gaussian Process (GP) | Small-scale affinity measurements (KD) | 0.65 - 0.80 | Provides uncertainty estimates. |
| Graph Neural Network (GNN) | Structural models of variants | 0.70 - 0.83 | Incorporates 3D structural information. |
Visualization Diagrams
Diagram Title: Integrated Directed Evolution & ML Workflow
Diagram Title: CRISPR-Yeast-ML Protocol Steps
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Item | Function in Integrated Workflow |
|---|---|
| CRISPR-Cas9 Plasmid (for host) | Expresses Cas9 nuclease and target-specific gRNA to create double-strand breaks for precise library integration (e.g., into yeast display locus). |
| Homology-Directed Repair (HDR) Donor Library | Linear DNA template containing the variant library flanked by homology arms (≥500 bp) for CRISPR-mediated integration, ensuring genomic stability. |
| M13KE Phagemid Vector | Allows fusion of protein variants to M13 phage pIII protein for phage display library creation from enriched pools. |
| Fluorescently-Labeled Antigen (for FACS) | Binds to displayed proteins on yeast surface, enabling quantitative sorting based on binding affinity and specificity via fluorescence intensity. |
| Magnetic Beads (Streptavidin) | Used for efficient immobilization of biotinylated target antigens during phage biopanning, facilitating rapid washing and elution steps. |
| Next-Generation Sequencing (NGS) Kit | For deep sequencing of pre- and post-selection pools to generate quantitative fitness data (enrichment scores) for machine learning training. |
| ML Feature Encoding Library (e.g., OneHot, AAindex) | Converts protein sequence data into numerical vectors suitable for model training (CNNs, Transformers). |
Conclusion
CRISPR-Cas mediated directed evolution represents a powerful synthesis of precision genome editing and evolutionary principles, offering researchers unprecedented control and speed in sculpting protein function. By mastering the foundational concepts, implementing robust methodological pipelines, proactively troubleshooting key challenges, and rigorously validating outcomes against benchmarks, scientists can harness this technology to solve complex problems in drug development and synthetic biology. The future points toward even more integrated systems—combining CRISPR libraries with advanced screening automation and AI-driven predictive models—to rapidly generate novel therapeutics, diagnostics, and biocatalysts, fundamentally accelerating the pace of biomedical innovation.